ФРИДЕЛЯ - КРАФТСА РЕАКЦИЯ
,
алкилирование и аци-лирование ароматич. углеводородов (аренов) и их производных
в присут. безводного AlCl3 и др. кислотных катализаторов. Классич.
пример Ф.- К. р.- алкилирование бензола ал-килгалогенидами, представляющее собой
типичное электроф. замещение в ароматич. ядре (SE):

В Ф.- К. р. вступают также
конденсир. ароматич. углеводороды и нек-рые гетероароматич. соед. (напр., тиофен,
фуран и их производные). Р-цию проводят в р-рителе или без него в небольшом
интервале т-р.
При алкилированиив качестве
реагентов используют алкилгалогениды, спирты и олефины; в качестве катализаторов
- к-ты Льюиса, протонные к-ты, кислотные оксиды, а также катионообменные смолы.
Катализаторы алкилирова-ния по общей активности можно примерно расположить в
след. ряды:
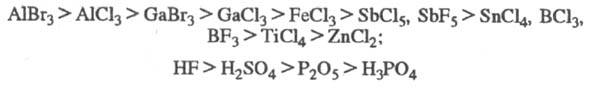
Активность катализаторов
в каждом конкретном случае зависит от природы субстрата, условий эксперимента
и наличия примесей, напр. следов воды.
Механизм р-ции алкилирования
предполагает, что сначала алкилгалогенид и катализатор образуют реагент (быстрая
стадия), представляющий собой комплекс с переносом заряда (ф-ла I) или ионную
пару (II), к-рый затем реагирует с ареном (медленная стадия), давая соед. III;
перенос протона к р-ри-телю приводит к продукту р-ции. При проведении процесса
без р-рителя или в слабоосновном р-рителе III - конечный продукт алкилирования,
он распадается лишь при разложении реакц. массы:
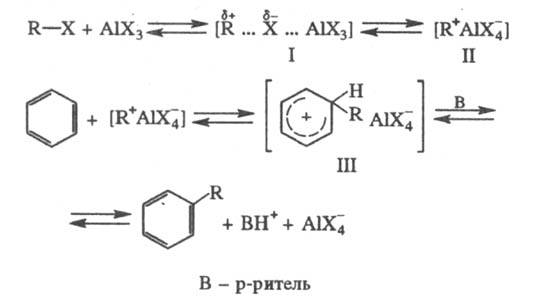
Реакц. способность алкилгалогенидов
в Ф.- К. р. падает при переходе от фторидов к иодидам (CH3Br реагирует
в ~200 раз быстрее, чем CH3I) и от третичного алкила к первичному.
Вводимая алкильная группа активирует бензольное ядро, что способствует дальнейшему
алкилированию. Число вводимых алкильных групп определяется стерич. факторами.
Для получения моноалкилир. продуктов используют избыток арена (последний может
служить р-рителем) и эффективное перемешивание.
Толуол алкилируется быстрее,
чем бензол. Соотношение орто-
и пара
-изомеров при алкилировании
толуола зависит от размера входящей алкильной группы; так, при алкилировании
CH3Br оно составляет (1,9:1), (СН3)2СНВг -(1,2:1),
C6H5CH2Cl- (0,82:1), при алкилировании трет-
бу-тилбромидом
(кат.- AlCl3) продукт орто
-замещения не обнаружен.
В большинстве случаев мета
-ориентирующие
заместители, напр. группа NO2, препятствуют алкилированию, однако
р-ция все же возможна, если молекула субстрата содержит одновременно активирующую
и дезактивирующую группы, как в случае о-нитроанизола.
Алкилирование олефинами
протекает в присут. безводного HCl с образованием активного комплекса:
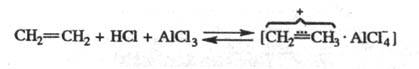
Кол-во катализатора в Ф.-
К. р. зависит от алкилирующего агента: при алкилировании алкилгалогенидами и
олефинами используют каталитич. кол-ва, спиртами и алкеноксидами -эквимолярные;
последнее условие должно соблюдаться при алкилировании аренов, содержащих в
качестве заместителей группы RO, RC(O) и др., способные образовывать комплексы
с AlCl3.
Побочные процессы при алкилировании
по Ф.- К. р.- изомеризация и диспропорционирование алкильных групп в арене.
Использование вместо AlCl3
катализаторов типа цеолитов позволяет повысить селективность Ф.- К. р. Так,
при алкилировании толуола этиленом в присут. AlCl3 - HCl образуется
смесь орто-
, мета-
и пара
-этилтолуолов в соотношении (11:55:34),
а при проведении процесса на модифицир. цеолите ZSM-5 получают 97% и-этилтолуола
и 3% мета
-изомера; орто
-изомер не образуется.
Ацилирование в условиях
Ф.- К.р. проводят с помощью хлорангидридов или ангидридов карболовых к-т. Механизм
р-ции аналогичен алкилированию: на первом этапе ацилирующий агент и катализатор,
к-рый в этих случаях правильнее называть реагентом, образуют ионную пару (IV),
последняя с ареном дает соед. (V); перенос протона к р-ри-телю приводит к устойчивому
комплексу (VI), из к-рого продукт р-ции освобождается при разложении реакц.
массы водой:
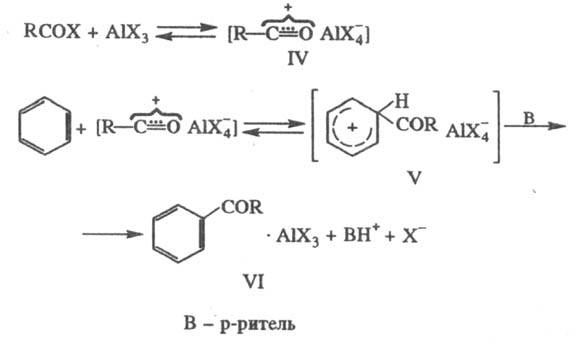
При ацилировании с помощью
ангидридов карбоновых к-т связываются два моля AlX3. Кол-во катализатора
в р-циях ацилирования, в отличие от р-ций алкилирования, должно
быть не менее, чем стехиометрическое;
это связано с тем, что как ацилирующий агент, так и продукт р-ции образуют с
катализатором комплексы в соотношении 1:1.
Влияние заместителей в
р-циях ацилирования такое же, как для р-ций алкилирования. Ацилирование
по Ф.- К. р., в отличие от алкилирования, приводит, как правило, к моноацильному
продукту.
Ацилирование в условиях
Ф.- К. р.- основной метод синтеза ароматич. и жирноароматич. кетонов; при использовании
хлорангидридов дикарбоновых к-т образуются дикетоны, напр.:
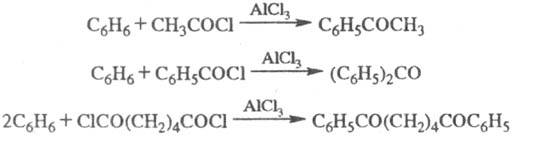
Частные случаи ацилирования
по Ф.- К. р.- Гаттермана -Коха реакция
, Фриса перегруппировка
. Модификация
Вильсмайера-Хаака: ацилиро-вание ароматич. соед. анилидами в присут. POCl3:
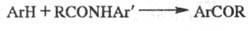
Модификация Бена: использование
в качестве р-ри-теля при ацилировании фенолов нитробензола для обеспечения гомогенности
реакц. смеси.
Модификация Ненцкого: ацилирование
фенолов карбоновыми к-тами в присут. ZnCl2; применение FeCl3
как более мягкого катализатора.
Модификация Перрье: ацилирование
с помощью предварительно приготовленного комплекса AlCl3 с RCOX с
целью повышения выхода продуктов р-ции.
Вариант Ф.- К. р.- метод
Фриделя - Крафтса - Каррера -получение нитрилов взаимод. ароматич. соед. со
свежеприготовленными галогенцианидами в присут. AlCl3:
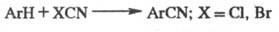
Ф.- К. р. применяют в пром-сти
для произ-ва высокооктановых топлив, изопропилбензола
, b-фенилэтилового спирта, бензофенона
, полупродуктов для лек. в-в и синтетич. красителей.
Р-ция открыта Ш. Фриделем
и Дж. Крафтсом в 1877.
Лит.: Tомас Ч.,
Безводный хлористый алюминий в органической химии, пер. с англ., M., 1949; Пrайс
Ч., в кн.: Органические реакции, пер. с англ., сб. 3, M., 1951; Берлинер Э.,
там же, сб. 5, M., 1951; Лебедев Н.Н., Химия и технология основного органического
и нефтехимического синтеза, 2 изд., M., 1975; Friedel-Crafts and related reactions,
ed. by G.A. Olah, v. 1-4, N.Y.-L., 1963-65. C.K. Смирнов.
|


