ГАЛОГЕНИРОВАНИЕ
(галоидирование), введение галогена в молекулу орг. соединения. Осуществляют
путем р-ций замещения (заместительное Г.) или присоединения (присоединительное
Г.).
Заместительное
галогенирование. При действии галогенов на насыщ. углеводороды (металепсия)
процесс протекает при инициировании светом по свободнорадикальному цепному
механизму, напр.:
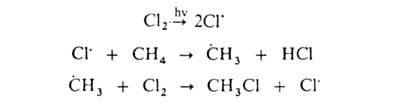
По свободнорадикальному
механизму идет также Г. углеводородных цепей жирноароматич. соединений.
В присут. к-т Льюиса р-ция протекает по электроф. механизму, напр.:
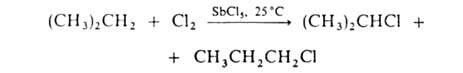
Г. алифатич.
карбоновых к-т в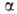 -положение
проводят с помощью С12 или Вг2 в присут. красного
Р (Гелля-Фолъгарда-Зелинского реакция). Замещение
-положение
проводят с помощью С12 или Вг2 в присут. красного
Р (Гелля-Фолъгарда-Зелинского реакция). Замещение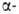 водородных
атомов в алифатич. и жирноароматич. карбонильных соед. идет через присоединение
галогена к енольной форме, напр.:
водородных
атомов в алифатич. и жирноароматич. карбонильных соед. идет через присоединение
галогена к енольной форме, напр.:
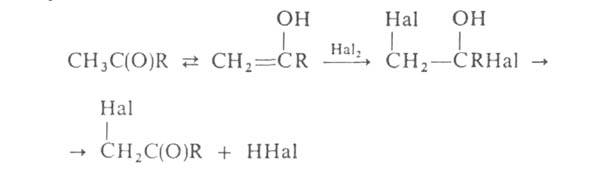
Действием
N-галогенамидов, гл. обр. N-бромсукцинимида, в присут. пероксидов осуществляют
свободнорадикальное Г. олефинов, жирноароматич. и гетероароматич. соед.
по метальной или метиленовой группе, соседней с двойной связью или циклом
(Воля - Циглера реакция
).
Замещение
атомов Н на F с образованием полифторзамешенных соед. проводят путем электрохим.
фторирования в безводном HF (р-ция Саймонса), действием CoF3
и др.
Заместительное
Г. в ядро ароматич. и гетероароматич. соед. протекает по механизму электроф.
замещения; обычно его осуществляют с использованием катализаторов (гл.
обр. апротонных или протонных к-т), напр.:
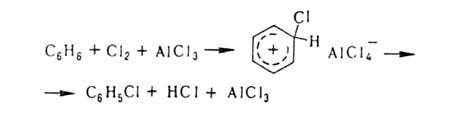
Если
в ядре этих соед. присутствуют пассивирующие заместители, процесс можно
проводить действием катиона галогена, образующегося из молекулярного галогена
и соли Ag в среде сильной протонной к-ты (р-ция Биркенбаха-Губо-Уотерса),
напр.:
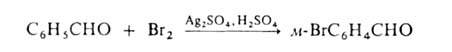
Замещение
на галоген атомов, отличных от водорода, или группы атомов осуществляется
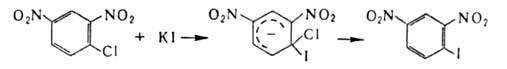
чаще всего по нуклеоф. механизму. В алифатич. соед. для замены атомов галогенов
(гл. обр. С1 или Вr) на иод используют Nal (Финкелъштапна реакция),
а
на фтор-SbF3 (р-ция Свартса). Группы ОН замещают на хлор или
бром действием соответствующих галогеноводородов, тригалогенидов или оксигалогенидов
фосфора, а также тионилгалогенидов, а на фтор-действием диэтил-1,1,2-трифтор-2-хлорэтиламина
или SF4. Карбоксильную группу замещают на С1, Вг или I действием
на серебряные соли карбоновых к-т соответствующего галогена (Бородина - Хунсдиккера реакция]. Заменой карбонильного кислорода в альдегидах
или кетонах на галоген (напр., с помощью РС15, PBr5,
SF4, MoF6) получают геминальные галогензамещенные.
В ароматич.
ряду для получения галогензамсщеиных используют замену групп NH2
на С1, Вr или I каталитич. разложением соответствующих солей диазония в
присут. порошка Сu (Гаттермана-Коха реакция
)или действием солей
Сu (Зандмепера реакция), а на F-разложением гидрофторидов диазония
(Шимана реакция
). Для Г. ароматич. и гетероароматич. соед. используют также
р-цию замещения (в т.ч. обмен галогенов), протекающую по механизму присоединения-отщепления
с промежут. образованием анионных комплексов,
напр.:
комплексов,
напр.:
Легкость
заместительного Г. действием галогенов уменьшается в ряду F " С1 > Вr >
I. Напр., для гомолитич. галогенирования метана до СН3На1 изменение
энтальпии в указанном ряду составляет соотв. -418, -105, -31 и + 54 кДж/моль.
Из-за высокой экзотермичности фторирование проводят при низких т-рах и
разбавлении F2 азотом, а чаще вместо F2 используют
фторсодержашие соединения. Иодирование действием I2, как правило,
идет с трудом и к тому же резко замедляется из-за обратимости р-ции. Поэтому
процесс ведут обычно в присут. окислителей (напр., HgO, HNO3),
окисляющих выделяющийся HI до I2, либо солей серебра, связывающих
анион I- в виде нерастворимой соли и одновременно обеспечивающих
генерирование Г.
Присоединительное
галогенирование. К ароматич. и гете-роароматич. соед. галоген присоединяется,
как правило, по радикальному механизму под действием света или при нагр.,
напр.:
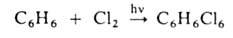
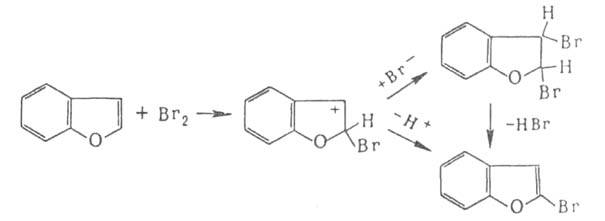
Если
цикл активирован, р-ция может протекать по ионному механизму, к-рый включает
стадию присоединения аниона галогена к промежуточно образующемуся в процессе
электроф. замещения комплексу,
напр.:
комплексу,
напр.:
Присоединение
галогенов по кратной связи происходит по электроф. или радикальному механизму.
Его можно осуществлять действием галогсноводородов (см. Гидрогалогенирование
),
межгалогенных
соед. (напр., ClBr, СП) или гипогалогенитов. В случае электроф. присоединения
может нарушаться правило Марковникова, что обусловлено образованием промежут.
мостикового катиона ф-лы I, напр.:
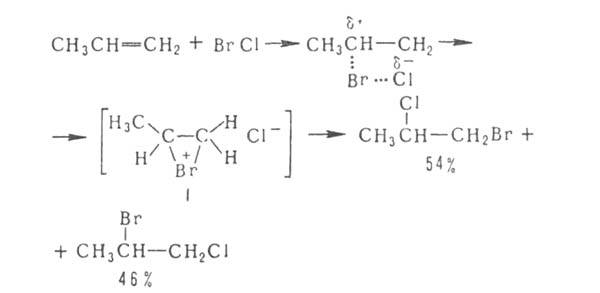
Способность
галогенов образовывать такие промежут. катионы возрастает в ряду: F " С1
< Вr < I. Образование мостековых ионов может приводить также к стереоселективному
присоединительному Г., причем способность заместителя при кратной связи
к стабилизации положит. заряда без образования мостикового иона снижает
стереоселективность. Так, при бромировании 1-фенил-1-пропена (в приведенном
ниже уравнении реакции R = Ph) образуется 88% эритро-изомера и 12% трео-изомера,
а в случае 1-(о-метоксифенил)- 1-пропена (R-o-CH3OC6H4)
- соотв. 63 и 37%:
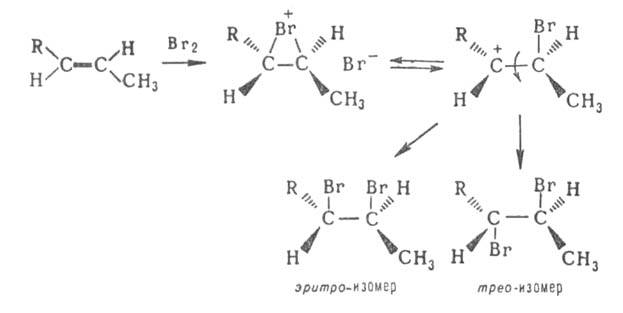
Присоединительное
Г. применяют для получения вицинальных и геминальных дигалогензамещенных,
напр.:

Для
введения в молекулу атома F широко используют варианты сопряженного присоединения
аниона F- и связанного с ним катиона по кратной связи в среде
HF, напр.:
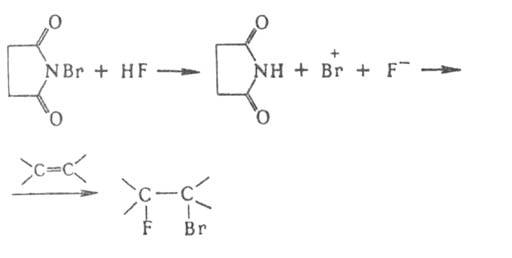
Иногда
Г. (заместительное и присоединительное) проводят действием галогеноводородов
(НС1 или НВr) и окислителя (т. наз. окислительное Г.). Р-цию осуществляют
в жидкой или газовой фазе (окислители соотв. Н2О2
и О2) в присут. соли Си на пемзе. См. также Галоформная реакция
,
Фторирование органических соединений.
Лит.:
Тереитьев
А. П., Яновская Л. А., в кн.: Реакции и методы исследования органических
соединений, кн. 6, М., 1957, с. 7-342; Кнунянц И. Л., Сокольский Г. А.,
там же, с. 343-87; Бюлер К., Пирсон Д., Органические синтезы, пер. с англ..
М., 1973, с. 374-468; Эфрос Л. С, Горелик М. В., Химия и технология промежуточных
продуктов. Л., 1980; Чамберс Р. Д., Джеймс С. Р, в кн.: Общая органическая
химия, пер. с англ., т. 1, М., 1981, с. 622-719. Л. И. Беленький


