ГОМОГЕННЫЙ КАТАЛИЗ
, ускорение хим. р-ции в присутствии катализатора,
к-рый находится в одной фазе с исходными реагентами (субстратами) в газовой
фазе или р-ре. При Г. к., как и при гетерогенном катализе
, катализатор
в р-ции не расходуется, однако является ее необходимым участником; без
катализатора р-ция протекает гораздо медленнее или не идет вовсе.
Механизм гомогенно-каталитических реакций. Можно выделить сравнительно
небольшую группу процессов, в к-рых участие катализатора не связано с образованием
определенного хим. соед. с субстратом. К таким процессам относится, напр.,
катализ парамагн. частицами синглет-триплетного превращения карбенов (изменяется
электронный спин молекулы) или орто-, пара-превращение Н2 (изменяется
ядерный спин). Формально к Г. к. можно отнести газофазные р-ции рекомбинации
атомов и простейших радикалов в присут. химически инертных частиц, к-рые,
участвуя в тройном соударении, обеспечивают отвод энергии, выделяющейся
при образовании хим. связи. Однако в огромном большинстве случаев механизм
Т.к., как и гетерог. катализа, включает более или менее сложное хим. превращ.
катализатора в р-циях.
Рассмотрим в кач-ве примера Г. к. окислит.-восстановит. некомплементарной
р-ции, т.е. такой р-ции, в к-рой число электронов, отдаваемых одной частицей
восстановителя и принимаемых одной частицей окислителя, не совпадает. В
р-ции 2Се4+ + Tl+ -> 2Се3 +
+ Tl3+ последовательное одноэлектронное окисление Т1+
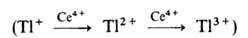
затруднено из-за высокого окислит.-восстановит. потенциала пары Т12+/Т1+
(для пары Т13+/Т12+ этот потенциал значительно ниже).
В присут. ионов Мп р-ция протекает по многостадийному механизму:
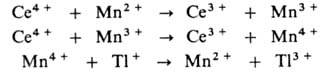
Ионы Мп переводят одноэлектронный перенос в р-циях Мп2+ и
Мn3 с Се4 + в двухэлектронный - в р-ции
Мn4+ с Т1+ (все р-ции с участием ионов Мп комплементарны).
Протекание всех стадий облегчается из-за того, что для пар Мn4+
/Мn3+ и Мn3+ /Мn2+ окислит.-восстановит.
потенциалы близки и расположены между значениями этих потенциалов для пар
Се4+ /Се3+ и Т13+ /Т1+ .
В радикально-цепных процессах в присут. нек-рых в-в может ускоряться
образование радикалов в р-циях зарождения или разветвления цепи. Так, при
окислении орг. в-ва RH в присут. ионов Со в небольшой концентрации ускоряется
разветвление цепи, а при высокой концентрации этих же ионов-также и развитие
цепи. Каталитич. механизм распада гидропероксида включает следующие стадии:
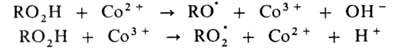
Развитие цепи каталитически ускоряется по схеме:
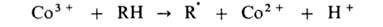
Эта же р-ция может дополнительно ускоряться в присут. ионов Вr-,
к-рые играют роль сокатализатора:
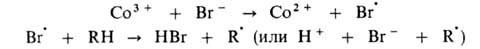
В кислотно-основном Г. к. под действием катализатора обычно усиливаются
электроф. или нуклеоф. св-ва молекул реагентов. К-ты и основания, ускоряющие
такие р-ции, могут служить катализаторами в недиссоциированной форме (общий
кислотно-основной катализ) либо воздействовать на субстрат ионами Н3О+
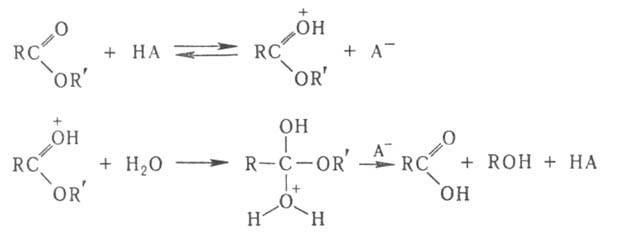
и ОН- (специфич. кислотно-основной катализ). Напр., при кислотном
гидролизе сложных эфиров каталитич. действие к-ты НА связано с протонированием
карбонильной группы, что облегчает последующее присоединение воды:
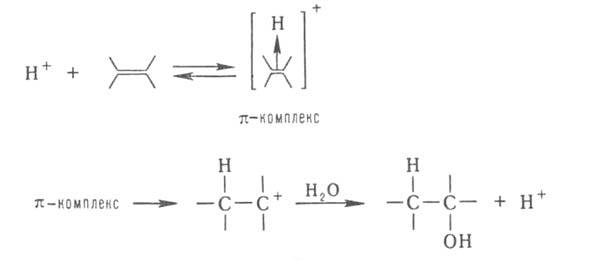
При гетеролитич. присоединении по кратным связям молекулы AD (А-акцепторная,
D-донорная часть молекулы) катализатором м. б. более сильный, чем AD, акцептор
(электрофил) А или донор (нуклеофил) D. Напр., при гидратировании олефинов
к-та Н+ служит катализатором, т. к. облегчает последующее взаимод.
с водой:
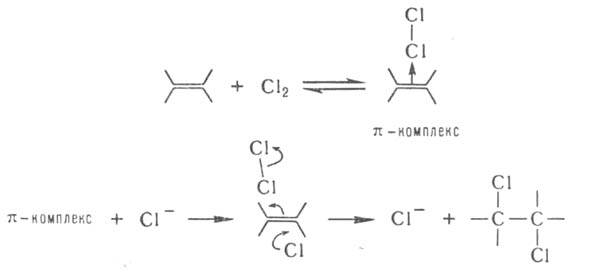
При присоединении Сl2 или НСl катализатором м. б. ион Сl-:
Апротонные к-ты могут катализировать гетеролитич. р-ции непосредственно
или образуя сильную протонную к-ту, если в системе присутствует вода или
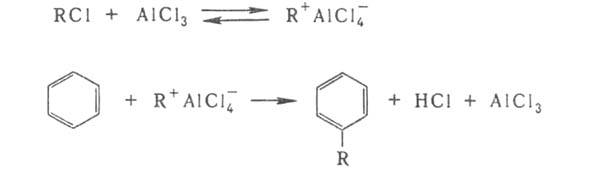
др. слабая протонная к-та. В р-ции Фриделя - Крафтса (алкилирование ароматич.
соед.) катализатором служит АlС13, усиливающий электроф. св-ва
алкильной группы в алкилгалогениде RC1:
Особенно высокой каталитич. активностью в гетеролитич. р-циях обладают
т. наз. бифункциональные катализаторы-соед., молекулы к-рых включают как
акцепторную, так и донорную группы. Р-ция в этом случае идет через циклич.
переходное состояние. Так, 2-гидроксипиридин - хороший катализатор мутаротации
глюкозы вследствие одноврем. участия в р-ции донорного атома N и акцепторной
группы ОН гидроксипиридина. Активность 2-гидроксипиридина существенно выше,
чем эквивалентной смеси пиридина и фенола, взятых в тех же концентрациях.
Усиление каталитич. эффекта по сравнению с монофункциональными катализаторами
определяется прежде всего энтропийным фактором-уменьшением числа частиц,
образующих активиров. комплекс.
В металлокомплексном Г. к. р-ции ускоряются в присут. комплексных соед.
Ti, Fe, Cu, Pt и др. переходных металлов, к-рые способны к образованию
комплексов с молекулами субстратов. Каталитич. активность м. б. обусловлена
след. факторами: 1) пространств.близостью молекул субстратов, входящих
как лиганды в координац. сферу металла, 2) ослаблением хим. связей в молекулах
субстратов и снижением энергии активации при их разрыве; 3) усилением вследствие
электронных эффектов донорных или акцепторных св-в молекул субстратов,
входящих в металлокомплекс; 4) снятием запретов по симметрии молекулярных
орбиталей благодаря участию d-орбиталей металлов; 5) возможностью многоэлектронных
р-ций с использованием d-электронов переходных металлов в некомплементарных
окислит.-восстановит. р-циях с субстратами, для к-рых последовательное
одноэлектронное окисление или восстановление термодинамически затруднено.
Комплексы переходных металлов, кроме того, иногда облегчают образование
своб. радикалов, что обеспечивает возможность катализа радикальных и радикально-цепных
р-ций. Металлокомплексные соед. катализируют гидрирование, окисление, полимеризацию,
карбонилирование олефинов и ацетиленов, фиксацию азота, диспропорционирование
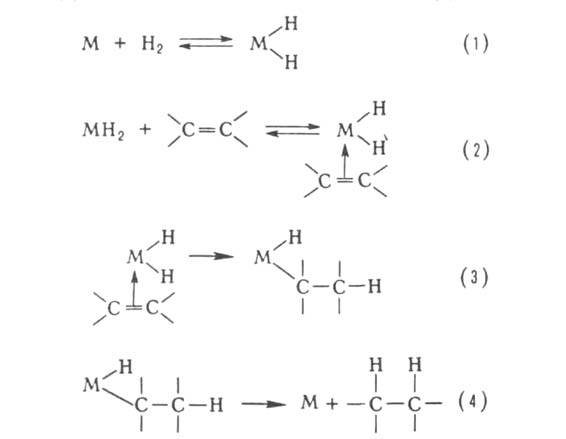
олефинов, активацию и разл. р-ции алканов и др. Типичный механизм металлокомплексного
Г. к. рассмотрен на примере каталитич. гидрирования олефинов. Он включает
стадии окислит. присоединения Н2 (1), образование комплекса
с олефином (2), внедрение молекулы олефина по связи М—Н (3), восстановит.
отщепление алкана (4):
Т. обр., во всех типах Г. к. катализатор, как правило, обеспечивает
новый хим. механизм процесса. Каталитич. р-ции происходят в неск. стадий,
в одной из к-рых частица катализатора входит в каталитич. цикл, а в другой
выделяется в своб. состоянии, чтобы вновь участвовать в р-ции. В этом смысле
гомогенно-каталитич. р-ции подобны цепным; их принципиальное отличие заключается
в том, что в цепных р-циях скорость образования продукта больше скорости
образования активного центра (скорости инициирования цепи) в v раз (v-
длина цепи), тогда как в каталитич. процессах скорость образования активного
центра катализатора больше скорости образования продукта или этот активный
центр присутствует в системе заранее.
Кинетика гомогенно-каталитич. р-ций определяется их механизмом (см. Каталитических реакций кинетика
).
Практическое применение. Г. к. используют для пром. получения:
спиртов-гидратацией олефинов в присут. к-т (H2SO4,
H3PO4); нитробензола и др. нитросоединений-нитрованием
ароматич. соед. в присут. H2SO4; уксусной к-ты-карбонилированием
метанола в присут. комплексов Rh (сокатализатор-HI); терефталевой к-ты-окислением
п-ксилола в присут. солей Со; альдегидов - гидроформилиро-ванием
олефинов в присут. карбонилов Со; лекарств, напр. L-ДОФА,-энантиоселективным
гидрированием аминокислоты в присут. комплекса Rh с хиральными лигандами
и др. Многие р-ции с участием металлокомплексных соед. по высокой регио-
и стереоселективности приближаются к реакциям ферментативного катализа.
Использование принципов действия ферментов позволяет разрабатывать принципиально
новые гомогенно-каталитические процессы.
Лит.: Гам мет Л., Основы физической органической химии, пер.
с англ., М., 1972, с. 407-45; Джен к с В., Катализ в химии и энзимологии,
пер. с англ., М., 1972; Литвиненко Л. М., Олейник Н. М., Органические катализаторы
и гомогенный катализ, К., 1981; Parshall G. W., Homogeneous catalysis,
N.Y., 1980. А. Е. Шилов.


