ХЮККЕЛЯ МЕТОД
, квантовохим. метод
приближенного расчета энергетич. уровней и мол. орбиталей ненасыщенных
орг. соединений. Основан на предположении, согласно к-рому движение электрона
вблизи атомного ядра в молекуле не зависит от состояний или числа др. электронов.
Это позволяет максимально упростить задачу определения мол. орбиталей (МО)
в представлении линейной комбинацией атомных орбиталей (см. ЛКАО-приближение
).
Метод предложен Э. Хюккелем в 1931 для
расчета электронного строения углеводородов с сопряженными связями. Считается,
что атомы углерода сопряженной системы лежат в одной плоскости, относительно
к-рой высшие занятые и низшие виртуальные (свободные) МО (граничные мол.
орбитали) антисимметричны, т. е. являются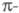 орбиталями,
образованными атомными 2рz-орбиталями (АО) соответствующих
атомов С. Влиянием остальных атомов, напр. Н, или мол. фрагментов с насыщенными
связями пренебрегают. Предполагается, что каждый из М атомов углерода
сопряженной системы вносит в
орбиталями,
образованными атомными 2рz-орбиталями (АО) соответствующих
атомов С. Влиянием остальных атомов, напр. Н, или мол. фрагментов с насыщенными
связями пренебрегают. Предполагается, что каждый из М атомов углерода
сопряженной системы вносит в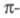 систему
один электрон и описывается одной атомной 2рz-орбиталью
систему
один электрон и описывается одной атомной 2рz-орбиталью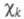 (k = 1, 2, ..., М). Каждая МО
(k = 1, 2, ..., М). Каждая МО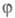 задается линейной комбинацией этих АО с коэф. С1, С2,
..., Cм;
задается линейной комбинацией этих АО с коэф. С1, С2,
..., Cм;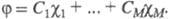 Значения коэф. определяются одноэлектронным ур-нием Шрёдингера [Шредингера], к-рое можно
представить в форме НС = ЕС, где Е -
одноэлектронная энергия;
С -вектор-столбец, составленный из коэф. Сk; H
- матрица одноэлектронного гамильтониана (см. Квантовая химия
). Матрица
H
характеризуется двумя параметрами - числами
Значения коэф. определяются одноэлектронным ур-нием Шрёдингера [Шредингера], к-рое можно
представить в форме НС = ЕС, где Е -
одноэлектронная энергия;
С -вектор-столбец, составленный из коэф. Сk; H
- матрица одноэлектронного гамильтониана (см. Квантовая химия
). Матрица
H
характеризуется двумя параметрами - числами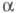 и
и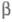 :
диагональные элементы матрицы для всех АО равны
:
диагональные элементы матрицы для всех АО равны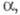 недиагональные элементы для пары АО химически связанных атомов равны
недиагональные элементы для пары АО химически связанных атомов равны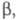 для всех остальных пар АО они равны нулю. Т. обр., матрица гамильтониана
однозначно указывает, какие из атомов химически связаны в соответствии
со структурной ф-лой в-ва. Важно, что вектор С (и, значит, МО
для всех остальных пар АО они равны нулю. Т. обр., матрица гамильтониана
однозначно указывает, какие из атомов химически связаны в соответствии
со структурной ф-лой в-ва. Важно, что вектор С (и, значит, МО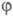 )
не зависит от выбора численных значений
)
не зависит от выбора численных значений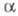 и
и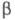 для
углеводорода. Поэтому в той мере, в какой сама структура матрицы Я передает
реальное строение молекулы и ее симметрию, X. м. позволяет дать качеств.
оценки относит. расположения уровней энергии, кратности их вырождения,
описать вклад электронов отдельной орбитали в электронную плотность
молекулы
и т. д. Напр., для циклич. полиенов X. м. предсказывает, что одноэлектронные
уровни энергии
для
углеводорода. Поэтому в той мере, в какой сама структура матрицы Я передает
реальное строение молекулы и ее симметрию, X. м. позволяет дать качеств.
оценки относит. расположения уровней энергии, кратности их вырождения,
описать вклад электронов отдельной орбитали в электронную плотность
молекулы
и т. д. Напр., для циклич. полиенов X. м. предсказывает, что одноэлектронные
уровни энергии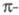 системы
расположены в след. порядке: низший из уровней невырожден, а несколько
следующих двукратно вырождены в силу аксиальной симметрии
системы
расположены в след. порядке: низший из уровней невырожден, а несколько
следующих двукратно вырождены в силу аксиальной симметрии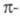 системы.
Наинизшее по энергии состояние молекулы соответствует полностью заполненным
электронным оболочкам, если число электронов равно 4п + 2, где п - любое целое число, что согласуется с правилом Xюккеля, согласно к-рому
такая полиеновая система ароматична (см. Ароматичность
). Более детальный
анализ симметрии и строения МО может объяснить, напр., природу диамагнетизма
ароматич. хим. соединений, четко проявляющуюся в спектрах ЯМР.
системы.
Наинизшее по энергии состояние молекулы соответствует полностью заполненным
электронным оболочкам, если число электронов равно 4п + 2, где п - любое целое число, что согласуется с правилом Xюккеля, согласно к-рому
такая полиеновая система ароматична (см. Ароматичность
). Более детальный
анализ симметрии и строения МО может объяснить, напр., природу диамагнетизма
ароматич. хим. соединений, четко проявляющуюся в спектрах ЯМР.
Простая модель электронного строения молекулы,
даваемая X. м., позволяет понять многие хим. явления. Напр., неполярность
альтернантных углеводородов
обусловлена тем, что эффективные заряды
на всех атомах углерода равны нулю. Напротив, неальтернантная конденсированная
система 5- и 7-членного циклов (азулен) имеет дипольный момент ок. 1Д (3,3
x 10-30 Кл x м).
В нечетных альтернантных углеводородах
основное энергетич. состояние отвечает электронной системе, в к-рой есть
хотя бы одна однократно занятая орбиталь. Можно показать, что энергия этой
орбитали та же, что и в свободном атоме, в связи с чем она наз. несвязывающей
МО. Удаление или добавление электрона изменяет заселенность лишь несвязывающей
орбитали, что влечет появление заряда на нек-рых атомах, к-рый пропорционален
квадрату соответствующего коэф. в разложении несвязывающей МО по АО. Для
определения такой МО применяют простое правило: сумма коэф.
Ck
для всех атомов, соседних с любым данным, должна быть равна нулю. Кроме
того, значения коэф. должны отвечать дополнит. условию нормировки: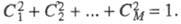 Это приводит к характерному чередованию (альтернированию) зарядов на атомах
в мол. ионах альтернантных углеводородов. В частности, указанное правило
объясняет выделение по хим. св-вам орто-
и пара
-положений
в бензольном ядре по сравнению с мета-
положением.
Это приводит к характерному чередованию (альтернированию) зарядов на атомах
в мол. ионах альтернантных углеводородов. В частности, указанное правило
объясняет выделение по хим. св-вам орто-
и пара
-положений
в бензольном ядре по сравнению с мета-
положением.
Закономерности, установленные в рамках
простого X. м., искажаются при более полном учете всех взаимод. в молекуле.
Однако обычно влияние множества разнородных дополнит, факторов (напр., электронов
остова, заместителей, межэлектронного отталкивания и т. п.) качественно
не меняет орбитальную картину электронного распределения. Поэтому X. м.
часто используют для моделирования сложных механизмов р-ций с участием
орг. соединений.
электронов
остова, заместителей, межэлектронного отталкивания и т. п.) качественно
не меняет орбитальную картину электронного распределения. Поэтому X. м.
часто используют для моделирования сложных механизмов р-ций с участием
орг. соединений.
При введении в молекулу гетероатомов (N,
О, S, ...) существенными становятся параметры матрицы H, принимаемые
для гетероатома и для атомов углерода. В отличие от случая полиенов, разные
типы атомов или связей описываются разными параметрами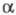 или
или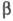 и
их соотношение существенно влияет на вид МО; качество предсказаний, получаемых
в рамках простого X. м., как правило, в итоге ухудшается.
и
их соотношение существенно влияет на вид МО; качество предсказаний, получаемых
в рамках простого X. м., как правило, в итоге ухудшается.
Простой по своей идее, наглядный и не
требующий сложных вычислений X. м. является одним из наиб. распространенных
средств создания квантовохим. модели электронного строения сложных мол.
систем. Наиб. эффективно его применение в тех случаях, когда св-ва молекулы
определяются в осн. топологич. структурой хим. связей, в частности симметрией молекулы
.
Попытки построить улучшенные варианты
X. м. в рамках простых молекулярных орбиталей методов
имеют мало
смысла, т. к. приводят к методикам расчета, сравнимым по сложности с более
точными методами квантовой химии.
Лит.: Стрейтвизер Э., Теория молекулярных
орбит. Для химиков-органиков, пер. с англ., М., 1965; Базилевский М.В.,
Метод молекулярных орбит и реакционная способность органических молекул,
М., 1969; Дьюap М., Догерти Р., Теория возмущений молекулярных орбиталей
в органической химии, пер. с англ., М., 1977.
В. И. Пупышев.
|


