КАТАЛИЗ
(от греч. katalysis - разрушение), изменение скорости хим. р-ции при воздействии в-в (катализаторов
), к-рые участвуют в р-ции, но не входят в состав продуктов. Катализатор не находится в стехиометрич. отношениях с продуктами и регенерируется после каждого цикла превращ. реагентов в продукты. Различают положительный и отрицательный К., в зависимости от того, ускоряет катализатор р-цию или замедляет ее. Как правило, термин "К." относят к ускорению р-ции; в-ва, замедляющие р-цию, наз. ингибиторами
. Каталитич. действие на р-цию могут оказывать образующиеся в ходе р-ции промежут. в-ва или продукты (см. Автокатализ
). Для К. характерно, что небольшие кол-ва катализатора ускоряют превращ. больших кол-в реагирующих в-в. Так, 1 мас. ч. Pt-катализатора вызывает превращ. 104 мас. ч. SO3 в SO2 или 106 мас. ч. NH3 в NO. Ускоряющее действие р-рителя на р-цию в р-рах обычно не относят к К. на том основании, что кол-во р-рителя, как правило, значительно превышает кол-во растворенных реагирующих в-в. Известны, однако, случаи ускорения р-ций в присут. очень малых добавок р-рителя, напр. воды.
Неизменность хим. состава и структуры катализатора по окончании процесса вряд ли может служить обязательным признаком К. Известно, что хим. состав выгруженного из реактора катализатора существенно иной, чем у загруженного; на состав и структуру катализатора влияет состав реакц. смеси. Неизменность хим. состава и структуры катализатора имеет смысл рассматривать по отношению к той из элементарных стадий сложной каталитич. р-ции, в к-рой непосредственно участвует катализатор, однако для этого необходимо надежно установить механизм р-ции, что не всегда возможно.
Термин "К." введен И. Берцелиусом в 1835.
При гомогенном катализе
катализатор и реагирующие в-ва находятся в одной фазе в молекулярно-дисперсном состоянии. При гетерогенном катализе
катализатор образует самостоят. фазу, отделенную границей раздела от фазы, в к-рой находятся реагирующие в-ва. Выделяют также гетерогенно-гомогенный К., при к-ром р-ция начинается на пов-сти твердого катализатора, а затем продолжается в объеме. Межфазным катализом
принято называть К. на границе двух несмешивающихся жидкостей; при этом роль катализатора состоит в переносе реагентов между фазами. Промежут. положение между гомогенным и гетерогенным К. занимает микрогетерогенный К. коллоидными частицами в жидкой фазе. Ускорение р-ций в присут. мицелл ПАВ наз. мицеллярным катализом. Исключительную роль в процессах в живых организмах играет ферментативный катализ
, обусловленный действием ферментов.
Важным компонентом пром. катализаторов являются промоторы -
в-ва, добавление к-рых к катализатору в малых кол-вах (проценты или доли процента) увеличивает его активность, селективность или устойчивость. Если промотор добавляется к катализатору в больших кол-вах или сам по себе каталитически активен, катализатор наз. смешанным. В-ва, воздействие к-рых на катализатор приводит к снижению его активности или полному прекращению каталитич. действия, наз. ядами каталитическими
. Встречаются случаи, когда одна и та же добавка к катализатору при одних концентрациях и т-рах является промотором, при других - ядом. В гетерог. К. широко применяют носители - в-ва, сами по себе каталитически неактивные или малоактивные. Нанесение на них катализатора значительно повышает его активность, гл. обр. вследствие увеличения пов-сти катализатора или предохранения его частиц от спекания (см. Нанесенные катализаторы
).
Общие закономерности катализа. Все каталитич. р-ции -самопроизвольные процессы, т. е. протекают в направлении убыли энергии Гиббса системы. Катализатор не смещает положения равновесия хим. р-ции, если не считать его влияния на коэф. активности реагирующих в-в в р-рах (или коэф. летучести, в случае р-ций в газовой фазе при высоких давлениях). Вблизи от равновесия один и тот же катализатор ускоряет прямую и обратную р-ции в равной степени, вдали от равновесия этого может и не быть. Из неск. возможных р-ций катализатор ускоряет не обязательно термодинамически наиб. выгодную, т.е. ту, для к-рой убыль энергии Гиббса максимальна. Напр., в присут. Bi2O3.MoO3 пропилен окисляется частично (до акролеина), в присут. Со3О4 происходит полное окисление (до СО2 и Н2О). Мерой селективности (избирательности действия) катализатора является отношение скорости vi р-ции, ведущей к накоплению i-го продукта, к суммарной скорости превращ. исходных в-в во всех возможных р-циях, т.е. vi/Svi. Расчет скорости каталитич. р-ции возможен на основе ряда моделей и приближений относительно ее механизма и режима протекания; для простейших случаев гомогенного и гетерогенного К. подход к расчету скорости изложен в ст. Каталитических реакций кинетика
.
Энергия активации Е каталитич. р-ции значительно меньше, чем для той же р-ции в отсутствие катализатора. Напр., для некаталитич. разложения NH3 на N2 + Н2 E ~ 320 кДж/моль, для того же разложения в присут. Pt Е ~ 150 кДж/моль. Благодаря снижению E обеспечивается ускорение каталитич. р-ций по сравнению с некаталитическими. Снижение E объясняется тем, что при К. р-ция протекает по новому механизму, складывающемуся из элементарных р-ций с меньшими энергиями активации, чем некаталитич. р-ция. При т. наз. стадийном механизме К. (кривая 1 на рис.) р-ция типа А : В (энергия активации E1) заменяется совокупностью стадий: 1) А + К : АК, 2) АК : В + К (энергии активации E2 и E3 соотв.), где К - катализатор, АК - устойчивое промежут. соед. реагента с катализатором. Для бимолекулярной р-ции А + В : С + D стадийный механизм может осуществляться по схеме: 1) А + К : АК, 2) АК + B : K + C + D. По такой схеме протекает, напр., окисление Н2 на металлич. катализаторе М: 1) М + 1/2O2 : МО, 2) МО + Н2 : М + Н2О. Одностадийные процессы К. (их наз. также ассоциативными или слитными) протекают по схеме: А + К : АК* : В + К. В этом случае (кривая 2 на рис.) катализатор не образует устойчивых промежут. соед. с реагентами, но входит в активир. комплекс АК*. Р-ция происходит с преодолением одного потенциального барьера, разделяющего начальное и конечное состояния системы, как и некаталитич. р-ция, но с пониженным значением энергии активации (E4 на рис.).
Большая скорость каталитич. р-ции м.б. обусловлена не только снижением энергии активации вследствие протекания р-ции по новому механизму, но и осуществлением под
действием катализатора цепного механизма р-ции. Напр., каталитич. действие паров воды на газофазное окисление СО объясняется образованием реакц. цепей с участием активных частиц Н и ОН. В гетерогенно-гомог. К. на пов-сти образуются активные частицы (напр., своб. радикалы НО2 и RO2 при окислении углеводородов RH), к-рые затем вылетают в газовую фазу и продолжают там цепь.
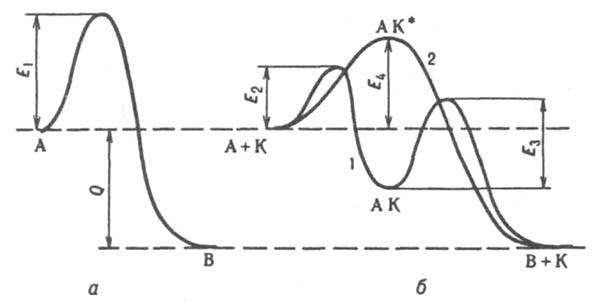
Энергетическая диаграмма реакции типа А : В; а - без катализатора, б - с катализатором К; 1 - при сталийном механизме, 2 - при ассоциативном механизме катализа: E1, E2, E4, E4 потенциальные барьеры, разделяющие исходное и конечное состояния системы, Q - тепловой эффект р-ции.
Доказано образование цепей на пов-сти катализатора при полимеризации олефинов и синтезе углеводородов из СО и Н2. Для мн. других гетерогенно-каталитич. р-ций обнаружены особенности, характерные для цепных р-ций: генерирование активных центров (чередование заполнения мест на пов-сти и их освобождение), образование активных промежут. частиц в сверхравновесных концентрациях (что способствует преодолению барьера энергетически невыгодных стадий), достижение макс. скорости р-ции спустя нек-рое время после ее начала. Каталитич. и цепные р-ции сближает также явление кинетич. сопряжения. Если превращ. катализатора при взаимод. с реагентами сопряжены с самой каталитич. р-цией (т. е. имеют общее промежут. в-во или активир. комплекс), становится возможным образование сверхравновесных концентраций активных центров на пов-сти катализатора и др. эффекты, типичные для цепных процессов.
Основные механизмы катализа. Каталитич. процессы, обусловленные переносом электрона (окисление, восстановление, гидрирование, дегидрирование, разложение нестойких кислородсодержащих соединений), относят к окислительно-восстановительному катализу
. Типичными катализаторами для них являются переходные металлы и их соед.: простые оксиды (V2O5, MnO2, МоО3, Сr2О3), шпинели (Fe3O4, CuCr2O4), сульфиды (MoS2, WS2) и др.; для р-ций в р-рах - соли и комплексные соед. переходных металлов. Высокая каталитич. активность этих в-в объясняется тем, что атомы переходных металлов могут существовать в разл. степенях окисления, изменение к-рых не требует больших энергетич. затрат. В результате перенос электрона от реагента к катализатору осуществляется легче, чем в отсутствие катализатора от восстановителя к окислителю. При одноэлектронном переходе образуются своб. радикалы, далее участвующие в р-ции. Напр., при переходе одного электрона от активного центра молибденового катализатора к кислороду образуется ион-радикал О2, участвующий далее в каталитич. окислении (Мо5+ + О2 : Мо6+ + О2; О2 + СnНm : продукт). Существует окислит.-восстановит. К. с многоэлектронным механизмом, при к-ром не образуются своб. радикалы в качестве промежут. частиц. Многоэлектронные переходы между катализатором и реагирующими молекулами возможны, если в активный центр катализатора входят неск. атомов переходного металла. Напр., в разложении Н2О2 активны комплексные соед., содержащие 2 иона Fe3+; в восстановлении мол. азота до N2H4 - комплексные соед., содержащие 2 или более ионов V2+.
К процессам кислотно-основного катализа
относятся каталитич. крекинг, гидратация, дегидратация, мн. р-ции изомеризации, конденсации орг. в-в. Типичные катализаторы для этого класса процессов - в-ва, способные передавать или принимать протон от реагентов или же способные к гетеролитич. взаимод. с реагентами (без разделения пары электронов). Среди этих в-в - протонные (H2SO4, CH3COOH, HF) и апротонные (BF3, AlCl3) к-ты, аморфные и кристаллич. алюмосиликаты, Аl2О3, фосфаты, сульфаты. Активными центрами в них является протонный центр Н+ (центр Брёнстеда [Бренстеда]) или акцептор электронной пары, напр., атом Аl (центр Льюиса). Реже применяются катализаторы основного характера (растворенные основания, твердые CaO, MgO и др.).
В случае т. наз. полифункциональных катализаторов отдельные этапы сложных каталитич. процессов окислит.-восстановительные и кислотно-основные - протекают на разных составных частях многокомпонентной многофазной системы. Напр., при неполном окислении непредельных альдегидов в непредельные к-ты в присут. оксидов Мо и V в элементарном акте происходят окислит.-восстановит. превращения катализатора:
V2O5 + СН2=СНСНО : СН2=СНСООН + V2O4;
2МоО3 + СН2=СНСНО : СН2=СНСООН + Мо2О5;
V2O4 + 1/2 О2 : V2O5, Mo2O5 + V2 O2 : 2МоО3
Конечная стадия р-ций - десорбция к-ты - происходит на пов-стях V2O5 и МоО3, обладающих слабокислотными св-вами. В водных р-рах каталитич. активность солей или комплексных соед. переходных металлов проявляется в определенном интервале рН. Это объясняется не только устойчивостью комплексных соед. при определенном рН, но и участием ионов Н+ и ОН- в элементарных стадиях К.
Катализ в промышленности. Несмотря на появление новых способов активации молекул (плазмохимия, радиац. химия, лазерная химия и др.), К. остается основой хим. произ-в. Относит. доля каталитич. процессов составляет 80-90% и продолжает возрастать; в общем объеме мирового пром. произ-ва каталитич. процессы дают ок. 18% стоимости всей продукции. В неорганическом синтезе важнейшими каталитич. процессами являются произ-во H2SO4, синтез NH3 из N2 и Н2, произ-во HNO3. В старейшем газофазном (нитрозном) способе произ-ва H2SO4 окисление SO2 в SO3 осуществлялось в присут. оксидов азота. В кон. 19 в. возник контактный процесс, при к-ром окисление SO2 в SO3 протекало в присут. Pt, нанесенной на разл. носители. Впоследствии Pt была заменена V2O5 с добавкой К2О и др. оксидов. Контактным способом получают десятки млн. т H2SO4 ежегодно.
Пром. синтез NH3 из N2 и Н2 был осуществлен в результате работ Ф. Габера и К. Боша в нач. 20 в. на железных катализаторах при давлениях ок. 300 атм и т-ре 450-500 °С. В настоящее время используют более активные Fe-катализаторы, промотированные V2O5, CaO, Аl2О3 и др. оксидами, что позволяет вести процесс при более низких давлениях и т-рах. Водород для синтеза NH3 получают путем двух последоват. каталитич. процессов: конверсии СН4 или др. углеводородов (СН4 + Н2О : СО + 3Н2) на Ni-катализаторах и конверсии образующегося оксида углерода (СО + Н2О : СО2 + Н2). Для достижения высоких степеней превращения последнюю р-цию осуществляют в две стадии: высокотемпературной (315-480°С) - на Fe-Cr-оксидных катализаторах и низкотемпературной (200-350°С) - на Cu-Zn-оксидных катализаторах. Hаиб. крупный потребитель NH3 - произ-во HNO3 окислением NH3 до NO на Pt и Pt-Rh сетках при 900-950 °С.
В органическом синтезе широкое применение К. началось в 1-й трети 20 в. благодаря работам П. Сабатье, В. Н. Ипатьева, Н. Д. Зелинского и др. Многочисл. р-ции гидрирования С=С,  , С=О, NO2-групп протекают на Ni-катализаторах, в числе к-рых Ni на носителях (кизельзуре, Аl2О3) и скелетный Ni - высокопористый катализатор, получаемый выщелачиванием Ni-Al сплавов. Реже применяют Сu, Со, Pt, Pd. К крупным пром. процессам относится
гидрогенизация жиров, превращ. бензола в циклогексан, нитробензола в анилин. В результате работ С. В. Лебедева и его учеников было создано произ-во синтетич. каучука. В его основе лежало получение мономера - бутадиена из этилового спирта по р-ции 2С2Н5ОН : С4Нб + 2Н2О + Н2 на смешанном оксидном катализаторе, сочетающем дегидратирующую, дегидрирующую и конденсирующую ф-ции, необходимые для всех стадий р-ции. Впоследствии мономеры в произ-ве синтетич. каучука - бутадиен, изопрен, стирол -стали получать каталитич. дегидрированием соответствующих парафинов и олефинов на Al-Cr-оксидных катализаторах. Началось пром. применение экономически еще более выгодного процесса получения мономеров окислит. дегидрированием на разл. оксидах переходных металлов (RCH2CH3 + 1/2O2 : RCH=CH2 + Н2О).
Широкое развитие в сер. 20 в. получили процессы каталитич. нефтепереработки; среди них - крекинг углеводородов нефти, для к-рого вначале основными катализаторами были аморфные алюмосиликаты, впоследствии цеолиты, отличающиеся более высокой активностью и большей селективностью по выходу парафиновых и ароматич. углеводородов. Для получения высококачеств. бензинов, дизельных и реактивных топлив применяют каталитич. риформинг, алкилирование, гидрокрекинг и гидроочистку. Катализаторы риформинга - Аl2О3, биметаллич. системы (Pt-Re на Аl2О3), реже оксиды Мо или Сr на Аl2О3; алкилирования - Н2SО4, HF, AlCl3, BF3; гидрокрекинга (переработки высококипящих фракций нефти под давлением Н2 в низкокипящие) - Аl-Со-Мо- и Al-Ni-W-системы. Близкие по составу катализаторы применяют в процессах гидроочистки, в к-рых под давлением Н2 тяжелые фракции нефти подвергаются обессериванию с выделением H2S; удаляются также азот- и кислородсодержащие соед. в результате гидрогенолиза соответствующих хим. связей. В условиях гидроочистки металлич. Ni-, Со-, Мо-, W-катализаторы превращаются в сульфиды (подробнее см. в статьях Каталитический крекинг
, Каталитический риформинг).
Каталитич. переработка угля в моторное топливо началась в 20-30-х гг. 20 в. в двух вариантах: прямая гидрогенизация угольной пасты и синтез углеводородов по Фишеру-Тропшу на Со- и Fe-содержащих катализаторах. После 2-й мировой войны в связи с быстрым развитием нефтепереработки эти процессы утратили свое значение, однако затем интерес к каталитич. переработке угля возобновился в связи с начавшимся истощением запасов нефти. Появились новые катализаторы, были созданы опытно-пром. и отдельные пром. установки. наиб. перспективен т. наз. Мобил-процесс, включающий газификацию угля, синтез метанола и послед. превращ. его в смесь углеводородов с большим выходом ароматич. углеводородов С8-С12 на высококремнистых цеолитах с сечением пор, приближающимся к поперечному размеру соответствующих ароматич. молекул.
К наиб. крупнотоннажным процессам каталитич. окисления относятся: окисление этилена в этиленоксид на серебряных катализаторах, окисление метанола в формальдегид на серебре или молибдате Fe, окисление пропилена в акролеин и окислит. аммонолиз пропилена с получением акрилонитрила на молибдате Bi. Высокая селективность последних двух процессов достигается за счет введения в катализатор оксидных добавок; применяют шести- и даже восьмикомпонентные оксидные катализаторы. Из гомог. жидкофазных процессов в пром-сти применяют окисление этилена в ацеталъдегид в водном р-ре, содержащем соли Сu и Pd, получение винилацетата окислением смеси С2Н4 и СН3СООН в присут. аналогичного катализатора и др. Каталитич. полимеризация получила развитие после открытия в 50-х гг. 20 в. К. Циглером и Дж. Наттой стереоспецифич. полимеризации олефинов на галогенидах, оксидах и др. соед. металлов IV-VIII групп (Ti, Zr, V, Сr, Мо и др.) с сокатализаторами - металлоорг. соед. Аl и нек-рых др. металлов I-III групп. В этих процессах получают кристаллич. полиолефины с регулярной структурой - полиэтилен,
полипропилен, полибутадиен и др. (подробнее см. в статьях Катализаторы окисления
, Катализаторы полимеризации
, Катализаторы процессов нефтепереработки
).
Каталитич. синтезы на основе СО быстро развиваются в связи с возрастающим значением ненефтяного сырья. Разработан пром. процесс получения уксусной к-ты карбонилированием метанола в присут. очень малых кол-в солей Rh. Быстро возрастает применение К. для очистки отходящих пром. газов доокислением вредных орг. примесей в СО2 на катализаторах глубокого окисления: металлах, простых оксидах (MnO2, Fe2O3), шпинелях (СuСr2О4, СоСr2О4) и др. Перспективна также разработка катализаторов, селективно удаляющих вредные серосодержащие примеси (H2S, SO2) из отходящих пром. газов и прир. газа. В 70-х гг. 20 в. возникло новое направление каталитич. очистки - удаление примесей из выхлопных газов автомобилей. Катализатор в дожигателях выхлопных газов должен доокислять примеси углеводородов и СО до СО2, а также восстанавливать оксиды азота до N2. Используют в дожигателях Pt, Pd, Rh, нанесенные на носители.
Лит.: Проблемы кинетики и катализа, т. 1-19, Л.-М., 1935-85; Боресков Г. К., Катализ, ч. 1-2, Новосиб., 1971; Томас Ч., Промышленные каталитические процессы и эффективные катализаторы, пер. с англ., М.. 1973; Гейтс Б., Кетцир Дж., Шуйт Г., Химия каталитических процессов, пер. с англ., М., 1981; Крылов О. В., "Кинетика и катализ", 1985, т. 26, № 2, с. 263-74; Advances in catalysis, v. 1-35, N.Y.-L, 1948-87. О. В. Крылов.
, С=О, NO2-групп протекают на Ni-катализаторах, в числе к-рых Ni на носителях (кизельзуре, Аl2О3) и скелетный Ni - высокопористый катализатор, получаемый выщелачиванием Ni-Al сплавов. Реже применяют Сu, Со, Pt, Pd. К крупным пром. процессам относится
гидрогенизация жиров, превращ. бензола в циклогексан, нитробензола в анилин. В результате работ С. В. Лебедева и его учеников было создано произ-во синтетич. каучука. В его основе лежало получение мономера - бутадиена из этилового спирта по р-ции 2С2Н5ОН : С4Нб + 2Н2О + Н2 на смешанном оксидном катализаторе, сочетающем дегидратирующую, дегидрирующую и конденсирующую ф-ции, необходимые для всех стадий р-ции. Впоследствии мономеры в произ-ве синтетич. каучука - бутадиен, изопрен, стирол -стали получать каталитич. дегидрированием соответствующих парафинов и олефинов на Al-Cr-оксидных катализаторах. Началось пром. применение экономически еще более выгодного процесса получения мономеров окислит. дегидрированием на разл. оксидах переходных металлов (RCH2CH3 + 1/2O2 : RCH=CH2 + Н2О).
Широкое развитие в сер. 20 в. получили процессы каталитич. нефтепереработки; среди них - крекинг углеводородов нефти, для к-рого вначале основными катализаторами были аморфные алюмосиликаты, впоследствии цеолиты, отличающиеся более высокой активностью и большей селективностью по выходу парафиновых и ароматич. углеводородов. Для получения высококачеств. бензинов, дизельных и реактивных топлив применяют каталитич. риформинг, алкилирование, гидрокрекинг и гидроочистку. Катализаторы риформинга - Аl2О3, биметаллич. системы (Pt-Re на Аl2О3), реже оксиды Мо или Сr на Аl2О3; алкилирования - Н2SО4, HF, AlCl3, BF3; гидрокрекинга (переработки высококипящих фракций нефти под давлением Н2 в низкокипящие) - Аl-Со-Мо- и Al-Ni-W-системы. Близкие по составу катализаторы применяют в процессах гидроочистки, в к-рых под давлением Н2 тяжелые фракции нефти подвергаются обессериванию с выделением H2S; удаляются также азот- и кислородсодержащие соед. в результате гидрогенолиза соответствующих хим. связей. В условиях гидроочистки металлич. Ni-, Со-, Мо-, W-катализаторы превращаются в сульфиды (подробнее см. в статьях Каталитический крекинг
, Каталитический риформинг).
Каталитич. переработка угля в моторное топливо началась в 20-30-х гг. 20 в. в двух вариантах: прямая гидрогенизация угольной пасты и синтез углеводородов по Фишеру-Тропшу на Со- и Fe-содержащих катализаторах. После 2-й мировой войны в связи с быстрым развитием нефтепереработки эти процессы утратили свое значение, однако затем интерес к каталитич. переработке угля возобновился в связи с начавшимся истощением запасов нефти. Появились новые катализаторы, были созданы опытно-пром. и отдельные пром. установки. наиб. перспективен т. наз. Мобил-процесс, включающий газификацию угля, синтез метанола и послед. превращ. его в смесь углеводородов с большим выходом ароматич. углеводородов С8-С12 на высококремнистых цеолитах с сечением пор, приближающимся к поперечному размеру соответствующих ароматич. молекул.
К наиб. крупнотоннажным процессам каталитич. окисления относятся: окисление этилена в этиленоксид на серебряных катализаторах, окисление метанола в формальдегид на серебре или молибдате Fe, окисление пропилена в акролеин и окислит. аммонолиз пропилена с получением акрилонитрила на молибдате Bi. Высокая селективность последних двух процессов достигается за счет введения в катализатор оксидных добавок; применяют шести- и даже восьмикомпонентные оксидные катализаторы. Из гомог. жидкофазных процессов в пром-сти применяют окисление этилена в ацеталъдегид в водном р-ре, содержащем соли Сu и Pd, получение винилацетата окислением смеси С2Н4 и СН3СООН в присут. аналогичного катализатора и др. Каталитич. полимеризация получила развитие после открытия в 50-х гг. 20 в. К. Циглером и Дж. Наттой стереоспецифич. полимеризации олефинов на галогенидах, оксидах и др. соед. металлов IV-VIII групп (Ti, Zr, V, Сr, Мо и др.) с сокатализаторами - металлоорг. соед. Аl и нек-рых др. металлов I-III групп. В этих процессах получают кристаллич. полиолефины с регулярной структурой - полиэтилен,
полипропилен, полибутадиен и др. (подробнее см. в статьях Катализаторы окисления
, Катализаторы полимеризации
, Катализаторы процессов нефтепереработки
).
Каталитич. синтезы на основе СО быстро развиваются в связи с возрастающим значением ненефтяного сырья. Разработан пром. процесс получения уксусной к-ты карбонилированием метанола в присут. очень малых кол-в солей Rh. Быстро возрастает применение К. для очистки отходящих пром. газов доокислением вредных орг. примесей в СО2 на катализаторах глубокого окисления: металлах, простых оксидах (MnO2, Fe2O3), шпинелях (СuСr2О4, СоСr2О4) и др. Перспективна также разработка катализаторов, селективно удаляющих вредные серосодержащие примеси (H2S, SO2) из отходящих пром. газов и прир. газа. В 70-х гг. 20 в. возникло новое направление каталитич. очистки - удаление примесей из выхлопных газов автомобилей. Катализатор в дожигателях выхлопных газов должен доокислять примеси углеводородов и СО до СО2, а также восстанавливать оксиды азота до N2. Используют в дожигателях Pt, Pd, Rh, нанесенные на носители.
Лит.: Проблемы кинетики и катализа, т. 1-19, Л.-М., 1935-85; Боресков Г. К., Катализ, ч. 1-2, Новосиб., 1971; Томас Ч., Промышленные каталитические процессы и эффективные катализаторы, пер. с англ., М.. 1973; Гейтс Б., Кетцир Дж., Шуйт Г., Химия каталитических процессов, пер. с англ., М., 1981; Крылов О. В., "Кинетика и катализ", 1985, т. 26, № 2, с. 263-74; Advances in catalysis, v. 1-35, N.Y.-L, 1948-87. О. В. Крылов.
|


