КЛЕТКИ ЭФФЕКТ
, общее назв. явлений, характерных для р-ций в жидкой и твердой фазах и обусловленных попаданием пары реагирующих частиц в окружение молекул среды ("клетку"). Типичные К.э. наблюдаются в р-рах при термич., фотохим. или радиац. распаде молекул RQR (константа скорости k1), в результате к-рого образуются радикальные пары RQ.R., окруженные молекулами р-рителя в той же полости, что и "материнские" молекулы. При этом происходит частичная рекомбинация радикалов с образованием исходных молекул RQR (константа скорости k-1):
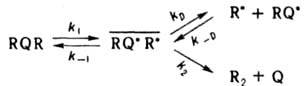
Остальные радикальные пары могут либо претерпеть превращ. с появлением продуктов Q и R2 (константа скорости k2), либо продиффундировать из клетки (константа скорости kD) и прореагировать с р-рителем SH или к.-л. реагентом в р-ре. Вышедшие из клетки радикалы, совершая независимые диффузионные движения, к-рые способствуют спиновой конверсии радикальных пар (см. ниже), могут с определенной вероятностью неоднократно возвращаться в исходную клетку (константа скорости k_D) и повторять все взаимод. до тех пор, пока не произойдет рекомбинация или р-ция одного из радикалов.
Внутриклеточная рекомбинация радикалов приводит к след. К.э.: 1) квантовый выход мономол. фотодиссоциации в р-рах значительно меньше, чем в газовой фазе, где он равен 1. Так, квантовый выход фотодиссоциации 12 и Вr2 в ССl4 составляет 0,14 и 0,22 соответственно. 2) При термич. или фотохим. распаде инициатора радикальной полимеризации наблюдаемая константа скорости kнабл существенно меньше ожидаемой k1, если k1, k2аk1. Если kD~0, то, согласно схеме, kнабл=klkD/(k1+kD+k2). 3) В системе появляются продукты внутриклеточной рекомбинации. Напр., при распаде ацетилпероксида в толуоле наряду с метаном, образующимся по р-ции 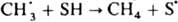 , появляется этан:
, появляется этан: 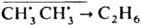 . При совместном распаде CH3N=NCH3 и CD3N=NCD3 в изооктане образуются только продукты внутриклеточной рекомбинации СН3 СН3 и CD3 CD3, a СН3 CD3 отсутствует. 4) При частичном распаде оптически активных молекул, напр. L L, на радикалы, имеющие своб. валентности на оптически активном атоме, могут образоваться молекулы LL, DD и LD, из-за чего происходит рацемизация оставшегося в-ва. 5) При частичном распаде меченых соед. на способные к изомеризации радикалы наблюдается внутримол. изотопный обмен. Напр., при
распаде ацилпероксидов, меченых по карбонилу 18О, метка переходит в пероксидную группу.
Др. группа К.э. связана с изменением суммарного электронного спина радикальной пары (спиновой конверсии). В частности, не способные к рекомбинации триплстные Т-пары превращ. в синглетныс S-пары, способные к внутриклеточной рекомбинации. Т. к. эти превращ. индуцируются магн. ядрами 13С, 15N, 17О, 19F, 31Р и др., в результате происходит обогащение этими изотопами продуктов внутриклеточной рекомбинации по сравнению с продуктами, образующимися из вышедших из клетки радикалов, входивших в состав немагнитных Т-пар-т. наз. изотопная селекция ядер, или магн. изотопный эффект (см. Магнитно-спиновые эффекты
). Напр., при фотодиссоциации бензоилпероксида в присут. триплетного сенсибилизатора в клетке оказывается Т-пара
. При совместном распаде CH3N=NCH3 и CD3N=NCD3 в изооктане образуются только продукты внутриклеточной рекомбинации СН3 СН3 и CD3 CD3, a СН3 CD3 отсутствует. 4) При частичном распаде оптически активных молекул, напр. L L, на радикалы, имеющие своб. валентности на оптически активном атоме, могут образоваться молекулы LL, DD и LD, из-за чего происходит рацемизация оставшегося в-ва. 5) При частичном распаде меченых соед. на способные к изомеризации радикалы наблюдается внутримол. изотопный обмен. Напр., при
распаде ацилпероксидов, меченых по карбонилу 18О, метка переходит в пероксидную группу.
Др. группа К.э. связана с изменением суммарного электронного спина радикальной пары (спиновой конверсии). В частности, не способные к рекомбинации триплстные Т-пары превращ. в синглетныс S-пары, способные к внутриклеточной рекомбинации. Т. к. эти превращ. индуцируются магн. ядрами 13С, 15N, 17О, 19F, 31Р и др., в результате происходит обогащение этими изотопами продуктов внутриклеточной рекомбинации по сравнению с продуктами, образующимися из вышедших из клетки радикалов, входивших в состав немагнитных Т-пар-т. наз. изотопная селекция ядер, или магн. изотопный эффект (см. Магнитно-спиновые эффекты
). Напр., при фотодиссоциации бензоилпероксида в присут. триплетного сенсибилизатора в клетке оказывается Т-пара 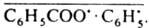 Поскольку в T-S-превращении преим. участвуют пары, содержащие 13С, кол-во этого изотопа в фенилбензоате, образовавшимся в результате внутриклеточной рекомбинации радикалов, на 23% больше, чем в бензоле, являющимся продуктом внеклеточных превращ.
Поскольку в T-S-превращении преим. участвуют пары, содержащие 13С, кол-во этого изотопа в фенилбензоате, образовавшимся в результате внутриклеточной рекомбинации радикалов, на 23% больше, чем в бензоле, являющимся продуктом внеклеточных превращ. 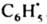 . К. э. проявляется также в том, что продукты, образующиеся при рекомбинации радикалов внутри клетки, и продукты превращения радикалов, вышедших из клетки, имеют противоположные знаки химической поляризации ядер.
К К. э. относят также снижение т. наз. связевой селективности углеводородов RH, напр., различие в скорости переноса атома Н от первичного, вторичного и третичного атома С к одному и тому же радикалу в жидкой фазе меньше, чем в газовой. Это объясняется конкуренцией между скоростью р-ции в клетке (внутриклеточного переноса Н) и скоростью выхода RH из клетки. В пределе, если скорость р-ции в клетке значительно превосходит скорость выхода из клетки, происходит практически полное нивелирование свя-зевом селективности. Напр., для р-ции RH+
. К. э. проявляется также в том, что продукты, образующиеся при рекомбинации радикалов внутри клетки, и продукты превращения радикалов, вышедших из клетки, имеют противоположные знаки химической поляризации ядер.
К К. э. относят также снижение т. наз. связевой селективности углеводородов RH, напр., различие в скорости переноса атома Н от первичного, вторичного и третичного атома С к одному и тому же радикалу в жидкой фазе меньше, чем в газовой. Это объясняется конкуренцией между скоростью р-ции в клетке (внутриклеточного переноса Н) и скоростью выхода RH из клетки. В пределе, если скорость р-ции в клетке значительно превосходит скорость выхода из клетки, происходит практически полное нивелирование свя-зевом селективности. Напр., для р-ции RH+ :
: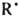 +Н2О найдено, что отношение констант скорости для метана и этана в газовой фазе составляет 1:30, а в водном р-ре -1:3. По указанной выше причине кинетический изотопный эффект
c=kH/kD для этой р-ции с участием С6Н12 и C6D12 в р-ре меньше, чем в газовой фазе. В жидкости, напр., cэфф=1.1, тогда как в газовой фазе cист~34. Еще большее нивелирование различий в реакц. способности обнаружено в случае р-ций в твердой фазе.
Лит.. Денисов Е. Т., Кинетика гомогенных химических реакций. М.. 1978; Беляков В. А.. Бучаченко А.Л., "Хим. физика", 1983. № II. с. 1510 14; ГрагеровИ.П.. Киприанова Л.А., Левит А.Ф.. Химическая поляризация ядер в исследовании механизма реакций органических соединений. К.. 1985: Лебедев Я.С. "Докл. АН СССР". 1985. т. 281. № 3. с. 636 40.
С. Г Энтелис
+Н2О найдено, что отношение констант скорости для метана и этана в газовой фазе составляет 1:30, а в водном р-ре -1:3. По указанной выше причине кинетический изотопный эффект
c=kH/kD для этой р-ции с участием С6Н12 и C6D12 в р-ре меньше, чем в газовой фазе. В жидкости, напр., cэфф=1.1, тогда как в газовой фазе cист~34. Еще большее нивелирование различий в реакц. способности обнаружено в случае р-ций в твердой фазе.
Лит.. Денисов Е. Т., Кинетика гомогенных химических реакций. М.. 1978; Беляков В. А.. Бучаченко А.Л., "Хим. физика", 1983. № II. с. 1510 14; ГрагеровИ.П.. Киприанова Л.А., Левит А.Ф.. Химическая поляризация ядер в исследовании механизма реакций органических соединений. К.. 1985: Лебедев Я.С. "Докл. АН СССР". 1985. т. 281. № 3. с. 636 40.
С. Г Энтелис
|


