КОРРОЗИЯ МЕТАЛЛОВ
(от позднелат. corrosio - разъедание), физ.-хим. взаимодействие металлич. материала и среды, приводящее к ухудшению эксплуатац. св-в материала, среды или техн. системы, частями к-рой они являются. В основе К. м. лежит хим. р-ция между материалом и средой или между их компонентами, протекающая на границе раздела фаз. Чаще всего это - окисление металла, напр.
3Fe+2О2=Fe3O4; Fe+H2SO4=FeSO4+Н2
По стехиометрии такие р-ции довольно просты, но по механизму они относятся к наиб. сложным гетерог. р-циям. Иногда при К. м. происходит и восстановление нек-рых компонентов материала; напр., при высоких давлениях и т-рах карбиды восстанавливаются в стали проникающим водородом. К К. м. нередко относят также нек-рые случаи их растворения в жидких металлах (напр., растворение сталей в жидкометаллич. теплоносителях ядерных реакторов).
К. м. - самопроизвольный процесс, сопровождающийся уменьшением энергии Гиббса системы конструкц. материал - среда. Для р-ций К. м. изменения энергии Гиббса по порядку величины таковы же, как и для самопроизвольно протекающих хим. р-ций. Термодинамич. нестабильность системы конструкц. металл - среда является причиной широкой распространенности К. м. во всех отраслях техники. Нормальная эксплуатация оборудования, коммуникаций, транспортных средств и т.п. часто возможна лишь при достаточном замедлении К. м., достигаемом при помощи многообразных способов и средств защиты от коррозии
. Изменением состава материала или среды или созданием особых условий можно добиться того, что К. м. самотормозится из-за образования поверхностных защитных слоев (см. Пассивность металлов
, Ингибиторы коррозии
, Коррозионностойкие материалы
). На нек-рых металлах (Al, Ti и др.) защитные слои в ряде сред образуются и без спец. мер.
Несмотря на успехи в борьбе с К. м., ее проблемы обостряются из-за непрерывного роста металлич. фонда и ужесточения условий эксплуатации металлов. Это связано с использованием высокоагрессивных сред (хим. пром-сть, ядерная и геотермальная области энергетики, разработка шельфа и др.), повышением рабочих т-р, давления и скоростей потоков, загрязнением атмосферы SO, и др.
примесями и т.п. Новые конструкц. материалы нередко подвергаются малоизученным и трудно прогнозируемым коррозионным разрушениям. Повышаются экологич. требования к средствам и технологиям защиты от коррозии; становится менее доступным сырье для произ-ва ряда коpрозионностойких материалов. В итоге возрастают как безвозвратные потери металла, стоимость к-рых входит в прямые убытки от К. м., так и затраты на защиту от коррозии. Однако наиб. велики косвенные убытки от К. м., связанные с простоями и снижением фондоотдачи, потерями и ухудшением качества продукции, авариями и т. п. В сумме косвенные и прямые убытки от К. м. и затраты на защиту [в соотношении примерно (3-4):1:1] в промышленно развитых странах достигают 4% национального дохода и более.
Механизм К. м. определяется прежде всего типом агрессивной среды. В сухих окислит. газах при повыш. т-рах на пов-сти большинства конструкц. металлов образуется слой твердых продуктов коррозии (окалина). При условии сплошности этого слоя скорость К. м. чаще всего лимитируется диффузией через него ионов металла к границе слой - газ или окислителя (напр., О2-) к границе слой-металл (подробнее см. Газовая коррозия
).
Иной механизм имеет очень распространенная К. м. в электролитич. средах - р-рах электролитов (в т.ч. в виде тонких пленок на пов-сти металла), пропитанных электролитами пористых и капиллярно-пористых телах (почвы, бетоны, нек-рые изоляц. материалы, рыхлые отложения и др.), а также в расплавах электролитов. В таких средах суммарный процесс К. м. можно записать в виде р-ции:
М+Ох=Мz++Red, (1)
где М - металл. Ох - частица окислителя. Red - его восстановл. форма (Ох имеет заряд +ze или Red - заряд -zе); здесь для упрощения принято равенство всех стехиометрич. коэффициентов. В преобладающем большинстве случаев р-ция (1) протекает по т. наз. электрохим. механизму: атом М и частица Ох непосредственно не контактируют, передача электронов от М к Ох происходит через зону проводимости М (рис. 1,а). Т. обр., процесс (1) фактически состоит из двух р-ций: анодного растворения
металла и катодного восстановления окислителя:
М=Mz++ze, (la) Ох+ze = Red. (16)
Скорость каждой из р-ций м. б. определена соответствующим кинетич. ур-нием (см. Электрохимическая кинетика
) и в этом смысле они полностью независимы, но при совместном протекании р-ции связаны условием электронейтральности системы. В нек-рых случаях возможно влияние продуктов одной р-ции на скорость другой.
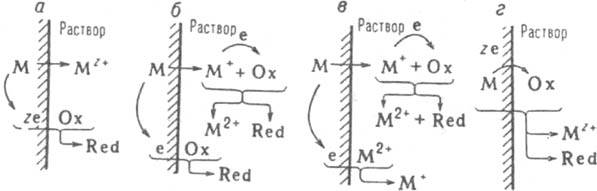
Рис. 1. Механизмы коррозии металлов: электрохимический (а); электрохимическо-химический (6); каталитический (в); предполагаемый химический (г). Для механизмов "6" и "в" принято зарядовое число z=2.
В электролитич. среде с высокой электрич. проводимостью ( металлич. пов-сть можно рассматривать как эквипотенциальную, т.е. имеющую одинаковый во всех точках электродный потенциал Е. Последний при стационарном протекании электрохим. К. м. принимает, как правило, определенное значение Eкор, при к-ром одинаковы скорости анодной и катодной р-ций, обычно выражаемые в
единицах плотности тока и обозначаемые ia и iк соответственно. Потенциал Eкoр наз. потенциалом коррозии или стационарным потенциалом; соответствующая ему величина плотности тока
ia=iк=iкор (2)
наз. скоростью или током коррозии. К. м. всегда необратимый процесс, поэтому значение Eкор не м. б. определено на основании термодинамич. соотношений и вычисляется только из кинетич. ур-ний р-ций (1а) и (16). В общем случае значения ia и iк зависят от потенциала E экспоненциально; эти зависимости в упрощенной записи имеют вид:
iа=kаехр(2,303Е/ba), (За) iк=kкcOxeхр(-2,З0ЗЕ/bк), (3б)
где cOx - концентрация окислителя Ох; ka и kк - эмпирич. постоянные при данной т-ре, к-рые, однако, могут зависеть от состава среды (kк не зависит от с0х); ba и bк - постоянные Тафеля (см. Тафеля уравнение
) для анодной и катодной р-ций соответственно. В координатах E-lgi зависимости (За) и (36) изображаются прямыми линиями (рис. 2, А). точке пересечения к-рых отвечают величины Екор и iкор. При этом из (За) и (36) следует:
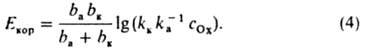
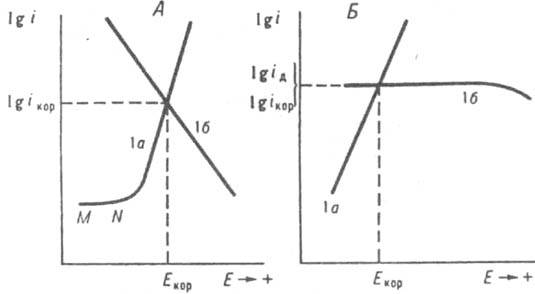
Рис. 2. Зависимость скорости i анодного растворения металла (l a) и катодного восстановления окислителя (1б) от электродного потенциала Е при элсюрохпм механизме коррозии. А - катодный процесс протекает в истино кинeтч. режиме. Б в режиме предельного диффузионного тока. iкор и Eкор значения тока и потенциала коррозии соотв.. iд - предельный диффузионный ток.
Ур-ния (За) и (36) отражают, в частности, кинетику типичной для неокислит. кислых сред электрохим. К. м. с восстановлением. Н+ - ионов; в водных р-рах р-ция (16) имеет вид: 2Н3О++2е=Н2+2Н2О. Если для анодной р-ции выполняется ур-ние (За), а скорость катодной р-ции полностью определяется диффузионным подводом Ох к пов-сти М, то величина iк максимальна в режиме предельного диффузионного тока iд (рис. 2, Б); в этом случае
ik=iд=iкор и Eкop=balg(iдka-1). (5) Соотношение (5) характерно для распространенной в нейтральных и нек-рых др. средах электрохим. коррозии с восстановлением растворенного кислорода, в водных р-рах р-ция (16) имеет вид:
О2+2Н2О+4е=4ОН-.
В большинстве случаев распределение на пов-сти металла точек мгновенного протекания р-ций (1а) и (1б) изменяется во времени статистически беспорядочно; соотв. средняя по времени скорость анодной р-ции (а значит. и скорость К. м.) в любой точке пов-сти одинакова и совпадает со скоростью катодной р-ции (равномерная или сплошная К. м.). Гетерогенность металла или среды, разл. условия подвода окислителя
или отвода продуктов коррозии, не нарушая эквипотенциальноети пов-сти (при высоких значениях (), могут приводить к возникновению на ней участков устойчивого предпочтительного протекания одной из р-ций - (1а) или (16), в соответствии с локальными значениями kа и ba, kк, bк и сOх (или iд). Для таких участков уже не выполняется равенство (2), т. с. ia№ik, а в предельном случае на одних участках со скоростью iа протекает практически только анодная р-ция, на других, со скоростью iк, - только катодная. Требование электронейтральности системы приводит к условию: iaSSa=iкSSк, где SS. и SSк - суммарные площади "анодных" и "катодных" участков соответственно. Чем больше значения iк и SSк/SSa, тем интенсивнее локальное растворение металла на анодных участках (в отсутствие пассивации).
При возрастании такой предпочтит. локализации, как правило, возрастает опасность локальных коррозионных разрушений, к-рая в реальных условиях чаще всего превосходит опасность для системы равномерной К. м. Причины описанной локальной К. м. многообразны: различия в составе зерна металла в объеме и на границе, концентрациях мех. напряжений, микровключения, разная природа контактирующих металлов, диффузионная неравнодоступность участков пов-сти и др. Участки пов-сти металла, на к-рых наблюдаются повыш. значения iа, м. б. макро- или микроскопическими. Первые наблюдаются обычно при контактной коррозии в месте соед. разнородных металлов, при щелевой коррозии (внутрищелевая пов-сть - анод, открытая - катод), на поздних стадиях питтинговой коррозии
(питтинги в виде крупных язв); вторые - при межкристаллитной коррозии
и на ранних стадиях питтинговой коррозии. Рост коррозионных трещин (см. Коррозия под напряжением
) в ряде случаев объясняют тем, что анодный процесс локализуется в вершине (острие) трещин.
В электролитич. среде с малой электрич. проводимостью ( за счет неоднородности металла или среды протяженная металлич. пов-сть м. б. неэквипотенциальной, т. е. для такой пов-сти характерно не одно значение Eкор, а нек-рое распределение потенциала. За исключением простейших по своей геометрии систем, теоретич. построение распределений потенциала и токов iа и iк при постоянстве ( требует решения дифференц. ур-ния Лапласа с разл. краевыми условиями. Однако кинетич. закономерности электрохим. К. м. и для такой пов-сти остаются справедливыми. Искомые распределения потенциала м. б. найдены указанным способом лишь при известных для каждого участка пов-сти значениях kа и bа, kк, bк и сOх (или iд). Осн. электрохим. механизм К. м., выражаемый ур-ниями (1а) и (16), может иметь варианты. Р-ция (1а) при z/2 может протекать через одноэлсктронныс стадии, напр. при z=2:
М=М++е, (6а)
М+=М2++е. (6б)
Катион промежут. валентности М+ в нск-рых случаях настолько устойчив, что может вступать в хим. р-цию
2М++Ох=2М2++Red (7)
прежде, чем успевает произойти его электрохим. анодное доокисление по р-ции (6б). Если при этом одновременно протекает р-ция (16), реализуется т. наз. электрохимическо-хим. механизм, при к-ром К. м. обусловлена электрохим. р-циями (6а) и (6б) и хим. р-цией (7) (рис. 1,6). Если на металлич. пов-сти вместо окислителя Ох, к-рый из-за р-ции (7) не достигает ее, восстанавливаются катионы М2+(М2++е=М+, рис. 1,в), осуществляется т. наз. каталитич. механизм К. м., при к-ром М+ играет роль катализатора р-ции (1). Эти варианты электрохим. механизма возможны в водных средах, но м. б. наиболее существенными при К. м. в орг. средах. Для таких сред, обычно характеризующихся малыми значениями (, ранее считалось обязательным протекание К. м. по т. наз.
хим. механизму, когда передача всех z электронов от М к Ох происходит непосредственно, в одном элементарном акте (рис. 1, г). В действительности же для электрохим. К. м. объемная величина х не имеет принципиального значения, по этому механизму протекает К. м. во многих малоэлектропроводных орг. средах; возможность хим. механизма сейчас допускают лишь для р-ров на основе неполярных р-рителей. В то же время в электропроводных водных р-рах (кислых и слабокислых) для ряда металлов при электродных потенциалах более отрицательных, чем Eкор (а при повыш. т-рах - и вблизи Eкор), скорость растворения не зависит от Е (участок MN на кривой а, рис. 2), причем этот эксперим. факт не м. б. объяснен диффузионными ограничениями. Одной из возможных причин его существования считают протекание р-ции (1) по хим. механизму.
Классификация К. м. определяется конкретными особенностями среды и условиями протекания процесса (подводом окислителя, агрегатным состоянием и отводом продуктов коррозии, возможностью пассивации металла и др.). Обычно выделяют К. м. в природных средах - атмосферную коррозию
, морскую коррозию
, подземную коррозию
, биокоррозию
; нередко особо рассматривают К. м. в пресных водах (речных и озерных), гсотeрмальных, пластовых, шахтных и др. Еще более многообразны виды К. м. в техн. средах; различают К. м. в к-тах (неокислительных и окислительных), щелочах, орг. средах (напр., смазочноохлаждающих жидкостях, маслах, пищ. продуктах и др.), бетоне, расплавах солей, оборотных и сточных водах и др. По условиям протекания наряду с контактной и щелевой К. м. выделяют коррозию по ватерлинии, коррозию в зонах обрызгивания, переменного смачивания, конденсации кислых паров; радиационную К. м., коррозию при теплопередаче, коррозию блуждающими токами и др. Особую группу образуют коррозионномех. разрушения, в к-рую входят помимо коррозионного растрескивания и коррозионной усталости фреттинг - коррозия
, водородное охрупчивание, эрозионная коррозия (в пульпах и суспензиях с истирающими твердыми частицами), кавитационная коррозия (при одноврем. воздействии агрессивной среды и кавитации). В общем случае воздействие агрессивной среды и мех. факторов на разрушение неаддитивно. Напр., при эрозионной К. м. потери металла вследствие разрушения защитной пленки м. 6. намного больше суммы потерь от эрозии и К. м. по отдельности.
Часто К. м. классифицируют также по отдельным металлам и их группам, по конкретным отраслям, произ-вам и объектам.
Коррозией часто наз. также происходящие при взаимод. со средами процессы разрушения неметаллич. материалов - полупроводников, бетона, полимеров, стеклопластиков и др. Представления о К. м., коррозионностойких материалах и защите от коррозии, коррозионных испытаниях
, проводимых при разработках и выборе материалов и ср-в защиты, выделяются в самостоят, научно-техн. дисциплину - химическое сопротивление материалов.
Лит.: Шлугeр М. А., Ажогин Ф. Ф., Ефимов М. А., Коррозия и зашита металлов, М., 1981; Коррозия. Справочник, под ред. Л. Л. Шрайера, пер. с англ., М.. 1981; Кeшe Г., Коррозия металлов, пер. с нем., М., 1984; Колотыркин Я. М., Металл и коррозия, М., 1985; Томашов Н. Д.. Чернова Г. П., Теория коррозии и коррозионно-стойкие конструкционные сплавы. М.. 1986. Л. И. Фрейман.
|


