ЛИОФИЛЬНОСТЬ И ЛИОФОБНОСТЬ
(от греч. lyo -растворяю, philed - люблю и phobos - страх), характеристики межмолекулярного взаимодействия
в-ва и среды (напр., воды, углеводорода), в к-рой оно находится. В случаях, когда средой служит вода, используют обычно термины "гидрофильность" и "гидрофобность" (от греч. hydor - вода), а если средой является углеводород (масло, жир) - термины "олео(или липо)фильность" и "олео(липо)фобность" (от лат. oleum и греч. lipos - масло).
Если в-во и среда близки по строению молекул или молекулы в-ва сильно взаимод. со средой, напр. образуют водородные связи
, то говорят о лиофильности, при слабом взаимод. в-ва и среды - о лиофобности. Мерой интенсивности межмол. взаимодействий может служить поверхностное натяжение
g пов-сти раздела в-ва со средой; степень лиофильности тем выше, чем ниже g. Количеств. критерии перехода от лиофобности к лиофильности, характеристики и эксперим. методы определения степени Л.
и л. зависят от строения изучаемой системы. Можно выделить: плоские пов-сти; частицы (порошки) и пористые тела; собственно дисперсные системы
, прежде всего свободнодисперсные. Понятия Л. и л. могут быть отнесены также к молекулам и их частям. Так, молекулы поверхностно-активных веществ
(ПАВ) образованы гидрофильными полярными группами и олеофильными углеводородными цепями; соотношение между этими частями молекул ПАВ определяет их гидрофильно-липофильный баланс (см. также Эмульсии
).
Л. и л. пов-сти твердого тела количественно характеризуется краевым углом смачивания
q (в воздушной среде); этот угол измеряется внутри жидкости (см., напр., рис., а). Пов-сть лиофильна по отношению к нанесенной на нее жидкости при q < 90°. При этом поверхностное натяжение границы раздела твердого тела с жидкостью меньше, чем границы раздела твердое тело - воздух, и работы адгезии Wa и когезии Wk связаны соотношением Wa > Wk/2. Пов-сть тем более лиофильна, чем ниже q; на предельно лиофильных пов-стях, когда Wa / Wk, происходит растекание жидкости. При q > 90° и Wa < Wk/2 пов-сть лиофобна. Гидрофильными являются, напр., пов-сти оксидов металлов, силикатных и алюмосиликатных минералов, гидрофобными - пов-сти парафина, фторопластов (см. подробнее Гидрофобное взаимодействие
).
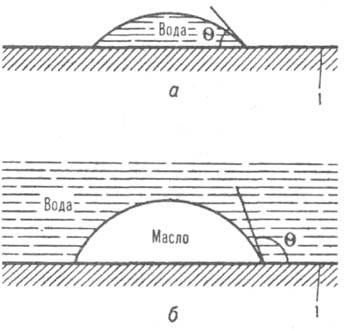
Смачивание водой гидрофильной - пов-ти (а) и избирательное смачивание oлeoфильной (гидрофобной) пов-ти (б); 1 - пов-сть твердого тела.
Углеводороды, имеющие низкую работу когезии, растекаются по большинству пов-стей (за исключением фторопластов), поэтому олеофобность и олеофильность м. б. охарактеризованы только по углу избирательного смачивания, к-рый измеряется при нанесении капли одной жидкости (напр., углеводорода) на пов-сть раздела твердого тела с другой жидкостью (водой; рис., б); угол q отсчитывается в более полярной жидкости (воде).
Л. и л. отдельных частиц можно характеризовать их способностью удерживаться пов-стью жидкости (см. Флотация
) или силами сцепления частиц при непосредств. контактировании в жидкости (см. Структурообразование
в дисперсных системах), а порошка или дисперсной пористой структуры - скоростью и равновесной высотой пропитки жидкостью. При высокой уд. пов-сти порошков и пористых тел Л. и л. могут быть охарактеризованы их способностью адсорбировать пары жидкости, теплотами адсорбции и смачивания или соотношением теплот смачивания двумя жидкостями. Тепловые эффекты особенно велики для твердых тел, способных к сольватации средой и набуханию в ней; напр., для крахмала отношение теплот смачивания водой и углеводородом может достигать 20. Степень лиофильности м. б. также охарактеризована величиной "исключенного объема" жидкости (по изменению р-римости в ней разл. в-в в присут. твердых частиц), по увеличению гидродинамич. радиуса движущихся частиц; более детальные сведения о взаимод. пов-сти твердого тела со средой дают спектральные методы.
Л. и л. дисперсных систем - понятия, к-рые первоначально использовались для описания обратимости коагуляции дисперсных систем и их чувствительности к
действию малых добавок электролитов; лиофильными коллоидами наз. р-ры высокомол. в-в. По совр. определению, к лиофильным коллоидным системам (ЛФЛКС) относят термодинамически равновесные микрогетерог. системы, образование к-рых из макроскопич. фаз сопровождается понижением термодинамич. потенциала и может идти самопроизвольно (самопроизвольное диспергирование). Лиофобные системы термодинамически неравновесны вследствие значит. избытка энергии на пов-сти раздела дисперсная фаза - дисперсионная среда.
Термодинамич. равновесность ЛФЛКС определяется компенсацией поверхностной энергии энтропийным фактором, связанным с участием частиц дисперсной фазы в тепловом (броуновском) движении. Такая компенсация осуществляется, если поверхностное натяжение межфазной пов-сти g12 меньше критич. величины gс = bkТ/d2, где k - постоянная Больцмана, Т - абс. т-ра, d - размер частиц, а коэф. b, зависящий от концентрации коллоидной системы, равен 5-10; при комнатной т-ре и d ~ 10-8 м, gс составляет сотые доли мДж/м2.
В ЛФЛКС реализуется равновесное распределение частиц по размерам. Узкое распределение частиц по размерам и высокое равновесное содержание в-ва в коллоиднодисперсном состоянии (высокая коллоидная р-римость) характерны для дисперсий мицеллoобразующих ПАВ (см. Мицеллообразование
); в этих дисперсиях уменьшение размеров частиц вызывает резкий рост поверхностного натяжения. Образование равновесной коллоидной системы при высокой мол. р-римости происходит в критич. системах.
Лиофобные дисперсные системы образуются в результате конденсации в-ва из гомог. фазы (напр., из пересыщенного р-ра) или при диспергировании макроскопич. фаз путем мех. измельчения либо электрораспыления, что требует значит. затрат энергии. Возможно диспергирование и без внеш. воздействия ("квазисамопроизвольное" диспергирование). Так, если поверхностная энергия границ зерен в поликристаллич. твердом теле больше или равна удвоенной энергии границы раздела твердое тело - жидкость (условие Гиббса - Смита), жидкая фаза распространяется по границам зерен с образованием дисперсной системы, в к-рой частицы разделены тонкими (10-7-10-8 м) жидкими прослойками.
Принципиальная термодинамич. неустойчивость лиофобных систем обусловливает протекание в них процессов, ведущих к изменению их строения и разрушению: коагуляции
, коалесценции (см. Эмульсии
, Пены
), диффузионному переносу в-ва от малых частиц к более крупным, седиментации. При высокой лиофильности частиц система м. б. термодинамически устойчивой к коагуляции; если при этом в-во дисперсной фазы нерастворимо в дисперсионной среде, св-ва дисперсии сходны со св-вами ЛФЛКС, в частности подобные "псевдолиофильные" системы могут возникать путем пептизации гелей и скоагулированных осадков, близкой по природе к самопроизвольному диспергированию.
Управление Л. и л. осуществляется хим. модифицированием пов-сти, напр. "прививкой" к пов-сти твердого тела лиофильных или лиофобных функц. групп, а также введением в систему ПАВ, изменяющих характер взаимод. на межфазной пов-сти. В зависимости от природы ПАВ возможна как частичная или полная лиофилизация, так и лиофобизация. Особенно эффективная лиофилизация достигается при образовании на пов-стях частиц т. наз. структурно-мех. барьера - плотных слоев низко- или высокомол. ПАВ, к-рые имеют лиофильную наружную пов-сть и способны противостоять мех. воздействию при приближении частиц друг к другу. Регулирование Л. и л. позволяет управлять св-вами пов-стей и дисперсных систем и широко используется во мн. областях техники и технологии.
Понятие о Л. и л. введено в 1909 Г. Фрёйндлихом [Фрейндлихом] как обобщение понятия "гидрофильность и гидрофобность", предложенного Ж. Перреном в 1905; большой вклад в развитие учения о Л. и л. внесен М. Фольмером,
П. А. Ребиндером, А. В. Думанским, Ф. Д. Овчаренко, Е. Д. Щукиным, А. И. Русановым и др.
Лит.: Сумм Б. Д., Горюнов Ю. В., Физико-химические основы смачивания и растекания, М., 1976; Физико-химическая механика и лиофильность дисперсных систем. Респ. межвед. сборник, К., в. 11, 1978; там же, 1981, в. 13; там же, 1985, в. 17; Щукин Е. Д., Перцов А. В., Амелина Е. А., Коллоидная химия, М., 1982. А. В. Перцов.
|


