МОЛЕКУЛЯРНЫХ ОРБИТАЛЕЙ МЕТОДЫ
, приближенные квантовохим. методы расчета волновых
ф-ций, энерге-тич. уровней и св-в молекул. Основаны на том, что для каждого
из электронных состояний молекулы как многоэлектронной системы полная волновая
ф-ция составляется из произведений волновых ф-ций электронов (мол. орбита-лей)
в соответствии с электронной конфигурацией системы, т. е. с учетом чисел заполнения
(1 или 2). Числа заполнения при
этом показывают, сколько электронов-один или два-занимают данную орбиталь, так
что данная орбиталь входит в произведение один или два раза. Поскольку, согласно
Паули принципу
, полная волновая ф-ция системы электронов должна быть
антисимметрична относительно перестановок индексов (номеров) электронов, ее
представляют в виде определителя (или линейной комбинации неск. определителей),
построенного из мол. спин-орбиталей и называемого о п р е д е л и т е л е м
С л е й т е р а (Слэтера).
Пусть, напр., система из
трех электронов имеет электронную конфигурацию (j1)2(j2)1,
где j1 и j2-мол. орбитали, а верх. индексы 1 и 2-числа
заполнения. Такой конфигурации отвечает набор спин-орбиталей j1a,
j1b, j2a, j2b, из к-рых м. б. построены две
волновые ф-ции системы в виде определителей Слейтера y1 и y2:

Число в скобках у спин-орбитали
указывает номер электрона, от пространств. переменных и спина к-рого зависит
эта спин-орбиталь. Множитель 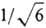 перед определителями выбран так, чтобы ф-ции y1 и y2 были
нормированы на единицу при нормированных и взаимно ортогональных мол. орбиталях
j1 и j2.
перед определителями выбран так, чтобы ф-ции y1 и y2 были
нормированы на единицу при нормированных и взаимно ортогональных мол. орбиталях
j1 и j2.
Волновые ф-ции в М. о.
м. обычно выбираются так, чтобы они отвечали т. наз. чистым спиновым состояниям,
т.е. были собств. ф-циями для операторов квадрата спина системы S2
и проекции спина на выбранную ось Sz. Так, записанные
выше ф-ции y1 и y2 являются собств. ф-циями для S2
с одним и тем же собств. значением 1/2 (1/2
+ 1)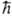 2
и для Sz с собств. значениями 1/2 ,
2
и для Sz с собств. значениями 1/2 , и — 1/2
и — 1/2 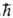 соотв. (
соотв. (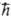 -постоянная
Планка). Как правило, основные состояния стабильных многоэлектронных систем
с четным числом электронов синглетны, т.е. отвечают собств. значениям операторов
S2 и Sz, равным нулю. В этом случае волновая
ф-ция системы м. б. представлена одним определителем, причем каждая мол. орбиталь
обязательно входит в него дважды: со спин-функцией а и со спин-функцией р, так
что число заполнения каждой мол. орбитали равно 2. Иначе говоря, у таких систем
имеется замкнутая электронная оболочка из двукратно заполненных мол. орбиталей.
Оболочкой при этом наз. совокупность орбиталей, вырожденных по к.-л. причине.
Напр., в случае многоэлектронного атома-это совокупность орбиталей с одним и
тем же главным и одним и тем же орбитальным квантовыми числами, но с разными
магнитным и спиновым квантовыми числами; замкнутой оболочкой обычно наз. как
полностью заполненную оболочку, так и все множество полностью заполненных оболочек.
Так, для атома Ne замкнутая оболочка (1s)2(2s)2(2p)6,
где 1s, 2s, 2p = {2рх, 2ру,
2рz}- символы атомных орбиталей, включает полностью
заполненные оболочки (1s), (2s)2 и (2р)6;
для молекулы Li2 в основном состоянии замкнутая оболочка (1sg)2
(1sи)2 (2sg)2, где 1sg,
1sи, 2sg-символы мол. орбиталей.
-постоянная
Планка). Как правило, основные состояния стабильных многоэлектронных систем
с четным числом электронов синглетны, т.е. отвечают собств. значениям операторов
S2 и Sz, равным нулю. В этом случае волновая
ф-ция системы м. б. представлена одним определителем, причем каждая мол. орбиталь
обязательно входит в него дважды: со спин-функцией а и со спин-функцией р, так
что число заполнения каждой мол. орбитали равно 2. Иначе говоря, у таких систем
имеется замкнутая электронная оболочка из двукратно заполненных мол. орбиталей.
Оболочкой при этом наз. совокупность орбиталей, вырожденных по к.-л. причине.
Напр., в случае многоэлектронного атома-это совокупность орбиталей с одним и
тем же главным и одним и тем же орбитальным квантовыми числами, но с разными
магнитным и спиновым квантовыми числами; замкнутой оболочкой обычно наз. как
полностью заполненную оболочку, так и все множество полностью заполненных оболочек.
Так, для атома Ne замкнутая оболочка (1s)2(2s)2(2p)6,
где 1s, 2s, 2p = {2рх, 2ру,
2рz}- символы атомных орбиталей, включает полностью
заполненные оболочки (1s), (2s)2 и (2р)6;
для молекулы Li2 в основном состоянии замкнутая оболочка (1sg)2
(1sи)2 (2sg)2, где 1sg,
1sи, 2sg-символы мол. орбиталей.
При построении волновых
ф-ций молекулы М. о. м. часто учитывают т. наз. условие симметрии: если конфигурация
ядер симметрична и при определенных операциях симметрии (поворотах, отражениях
в плоскости и др.) остается без изменений, то многоэлектронная волновая ф-ция
должна при таких преобразованиях меняться с учетом этой симметрии (другими словами,
преобразовываться по одному из неприводимых представлений той точечной группы,
операции симметрии к-рой оставляют конфигурацию ядер без изменений). Двухатомные
молекулы всегда обладают осевой симметрией, тогда как для многоатомных молекул
симметрия отсутствует, как только ядерная конфигурация претерпевает несимметричное
смещение от симметричной конфигурации.
Равновесные ядерные конфигурации часто обладают определенной симметрией и для
них М. о. м. требуют соблюдения условий симметрии. Конкретные проявления условий
симметрии состоят в том, что для невырожденных электронных состояний молекулы
мол. орбита-ли, из к-рых составляют определители, всегда м. б. выбраны так,
чтобы они преобразовывались по неприводимым представлениям точечной группы симметрии
ядерной конфигурации. В этих случаях говорят, что мол. орбитали относятся к
определенным типам симметрии (см. Симметрия молекул
).
Различие вариантов М. о.
м. определяется теми дополнит. требованиями, к-рые вводятся при поиске оптимальных
мол. орбиталей. В самом общем случае эти орбитали выбирают так, чтобы удовлетворялся
лишь вариационный принцип квантовой механики (см. Вариационный метод
).
Метод Хартри-Фока (метод
самосогласованного поля).
Орбитали ji,
отвечающие миним. значению энергии Е мол. системы, удовлетворяют уравнениям
Хартри-Фока, каждое из к-рых представляет собой одноэлектронное ур-ние типа
ур-ния Шрёдингера [Шредингера] с нек-рым эффективным одноэлектронным оператором, наз. фокианом
(обозначается F). В простейшем случае, когда число электронов N четное
и все орбитали ji(i = 1, 2, ..., N/2) дважды заняты,
ур-ния Хартри-Фока имеют вид:
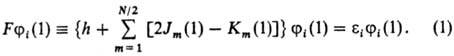
Фокиан F имеет смысл
оператора Гамильтона для электрона 1, находящегося в поле ядер и усредненном
поле всех остальных электронов молекулы. Он состоит из одноэлект-ронного оператора
h, равного сумме оператора кинетич. энергии электрона 1 и оператора потенц.
энергии его взаи-мод. со всеми ядрами, а также из суммы операторов (2Jm
— Km), определяющих взаимод. рассматриваемого электрона 1
с усредненным полем остальных электронов. Действие операторов Jm
и Кт на мол. орбиталь ji определяется
соотношениями
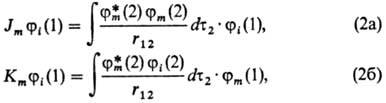
причем r12
есть расстояние между электроном 1 и к.-л. из остальных электронов, по переменным
к-рого проводится интегрирование (dx2dy2dz2
= dt2). Ф-ция jm
принадлежит к набору занятых орбиталей ji а jm*-комплексно
ей сопряженная. Интеграл в соотношении (2а) имеет смысл кулонов-ского потенциала
в точке, где находится электрон 1, создаваемого усредненным по всему пространству
распределением заряда электрона 2 с плотностью rm (2) = jm*
(2) jm (2). По этой причине операторы Jm наз.
кулоновскими. Интеграл в соотношении (2б) сложнее по своей структуре: он получается,
если в первом соотношении произвести обмен местами ф-ций jm
и ji с одновременной заменой их аргументов. Операторы Кт
наз. о б м е н н ы м и.
Величины ei
фигурирующие в (1) как собственные значения фокиана F, носят назв. орбитальных
энергий. Они определяют вертикальные потенциалы ионизации исходной мол. системы
в рассматриваемом приближении (см. Купман-са теорема). Орбитальные энергии
широко используются при интерпретации фото- и рентгеноэлектронных спектров,
в к-рых каждая полоса примерно отвечает потенциалу ионизации при удалении электрона
с той или иной мол. орбитали. Орбитали ji-, получаемые при
решении ур-ний Хартри-Фока, обычно наз. каноническими. Те из них, к-рые используются
при построении многоэлектронной волновой ф-ции, т. е. входят в нее с числами
заполнения 1 или 2, наз. з а н я т ы м и о р б и т а л я м и. Те же, к-рые не
используются при конструировании волновой ф-ции данного состояния системы, наз.
в и р т у а л ь н ы м и (свободными).
С помощью занятых орбиталей
определяется не только полная многоэлектронная волновая ф-ция, но и все характеристики
молекулы, к-рые м. б. вычислены с этой ф-цией. В частности, электронная плотность
в точке с радиусом-вектором r равна:
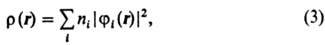
где ni-числа
заполнения орбиталей ji. Спиновая плотность
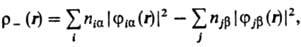
где пia
и njb-числа заполнения орбиталей jia
и jjb, входящих в волновую ф-цию со спин-функциями
a и b соответственно. Аналогичные соотношения можно написать и для др. величин
(дипольного момента молекулы, поляризуе-. мости и т.п.).
Решают ур-ния Хартри-Фока,
напр. ур-ния типа (1), обычно итерационным путем: выбирают на основе к.-л. соображений
начальные ф-ции (нулевое приближение) j0i, с ними
определяют операторы J0m и К0m,
затем решают ур-ния Хартри-Фока (1) и находят ф-ции ji(1)
первого приближения, исходные для след. шага итераций. Если в итоге получают
одни и те же ф-ции как под символами интегралов в операторах Jm
и Кт, так и в качестве решений (итерации сходятся), то
на этом расчет заканчивается. Решение ур-ний на конечном шаге итераций является
"согласованным с полем потенциала", к-рое определяется кулоновскими
и обменными операторами. Такое поле получило назв. с а м ос о г л а с о в а
н н о г о, а сам метод Хартри-Фока (во всем многообразии его вариантов)-м е
т о д а с а м о с о г л а с о в а нн о г о п о л я (ССП).
Метод самосогласованного
поля с молекулярными орбиталями в форме линейной комбинации атомных орбиталей.
Для молекулы указанная
итерационная процедура оказывается весьма трудоемкой. Задачу упрощают введением
для орбиталей ji приближенного представления в виде линейной
комбинации тех или иных базисных ф-ций cv (v = 1, 2,
..., М):
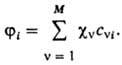
В этом случае неизвестными
оказываются лишь численные коэффициенты cvi. Обычно в качестве
известных базисных ф-ций выбирают атомные орбитали, каждая из к-рых центрирована
на к.-л. из ядер молекулы. Получаемые на основе вариационного принципа ур-ния
для определения коэффициентов cvi аналогичны ур-ниям Хартри
- Фока (и носят обычно то же самое название), однако с вычислит. точки зрения
они гораздо проще. При итерационном решении на каждом шаге итераций они сводятся
к системе линейных однородных ур-ний относительно cvi как
неизвестных величин, тогда как коэффициентами служат величины
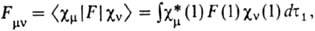
наз. м а т р и ч н ы м
и э л е м е н т а м и ф о к и а н а, а также интегралы
перекрывания
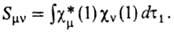
Для всех величин, характеризующих
мол. систему, напр. для ее полной энергии, дипольного момента, получаются выражения,
содержащие коэффициенты cvi. Так, для электронной плотности
получается вместо (3) след. выражение:
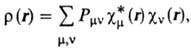
где 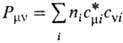 . Если рассматривать область пространства
вблизи данного ядра А, то электронная плотность в этой области
будет в осн. пропорциональна величинам Рmm при условии, что
ф-ция cm(r) центрирована у этого ядра. Поэтому величины
Рmm могут служить мерой электронной плотности, соотносимой
с орбиталями cm y ядра А, а величины SPmm при суммировании
по индексам орбиталей, центрированных у определенного ядра, могут служить э
ф ф е к т и вн ы м и э л е к т р о н н ы м и з а р я д а м и на атомах в молекулах.
Аналогично для атомов, между к-рыми имеется хим.
связь, величины SPmv (m относится к ядру
А, v-к ядру В) пропорциональны
электронной плотности в области между ядрами.
По этой причине Pmv часто наз. п о р я д
к а м и с в я з е
й. Сопоставление зарядов на атомах в разл. молекулах, а
также порядков связей между одними и теми же парами атомов служит для выяснения
качеств. картины перераспределения электронной плотности при переходе от одной
молекулы к другой, а также при изменении состояния молекулы. В зависимости от
конкретной задачи бывают удобны и другие определения зарядов на атомах и порядков
связей (напр., заселенности по Малликену, индексы Вайберга).
. Если рассматривать область пространства
вблизи данного ядра А, то электронная плотность в этой области
будет в осн. пропорциональна величинам Рmm при условии, что
ф-ция cm(r) центрирована у этого ядра. Поэтому величины
Рmm могут служить мерой электронной плотности, соотносимой
с орбиталями cm y ядра А, а величины SPmm при суммировании
по индексам орбиталей, центрированных у определенного ядра, могут служить э
ф ф е к т и вн ы м и э л е к т р о н н ы м и з а р я д а м и на атомах в молекулах.
Аналогично для атомов, между к-рыми имеется хим.
связь, величины SPmv (m относится к ядру
А, v-к ядру В) пропорциональны
электронной плотности в области между ядрами.
По этой причине Pmv часто наз. п о р я д
к а м и с в я з е
й. Сопоставление зарядов на атомах в разл. молекулах, а
также порядков связей между одними и теми же парами атомов служит для выяснения
качеств. картины перераспределения электронной плотности при переходе от одной
молекулы к другой, а также при изменении состояния молекулы. В зависимости от
конкретной задачи бывают удобны и другие определения зарядов на атомах и порядков
связей (напр., заселенности по Малликену, индексы Вайберга).
При использовании в качестве
базисных ф-ций атомных орбиталей метод Хартри-Фока часто наз. методом самосогласованного
поля с мол. орбиталями в форме линейной комбинации атомных орбиталей (ССП МО
ЛКАО) (т. е. для мол. орбиталей вводится ЛКАО-приближепиё [ЛКАО-приближепие]). Именно этот
вариант метода Хартри-Фока является пока основным при квантовохим. изучении
многоатомных молекул. Его достоинства - простота интерпретации и наглядность
получаемых результатов. Существ. недостаток - большое число подлежащих вычислению
мол. интегралов; их число увеличивается пропорционально М4,
где М-число базисных ф-ций (размерность базиса). Чтобы облегчить вычисления,
вводят ряд дополнит. предположений, имеющих, как правило, четкое физ. обоснование.
Однако эти предположения обычно настолько сильно упрощают задачу, что сохраняется
лишь общая структура ее решения в хартри-фо-ковском приближении. Для получения
количеств. заключений эти упрощения приходится хотя бы частично компенсировать
тем, что отдельные мол. интегралы либо их комбинации не вычисляются, а принимаются
за параметры. Значения этих параметров определяют для нек-рых молекул по набору
эксперим. данных (напр., энтальпий образования, частот квантовых переходов),
после чего их используют в качестве известных величин при расчетах др. молекул.
Такие М. о. м. наз. полуэмпирическими методами
.
Корреляционные диаграммы.
Орбитальная симметрия. Качеств. анализ системы мол. орбиталей и орбитальных
энергий на основе М. о. м. позволил сформулировать ряд фундам. положений соврем.
теоретич. химии. При непрерывном изменении параметров геом. конфигурации молекул
мол. орбитали и орбитальные энергии также меняются непрерывно. Это позволяет
построить т. наз. к о р р е л яц и о н н ы е д и а г р а м м ы, показывающие,
как меняются, напр., орбитальные энергии по мере сближения атомов, образующих
молекулу, от больших до равнoвесных межъядерных расстояний и далее-до слияния
ядер в единую систему с суммарным зарядом, отвечающим объединенному атому. Можно
также проследить, как меняются мол. орбитали и гибридизация атомных орбиталей
,
а также орбитальные энергии и числа заполнения орбиталей при изменении валентного
угла в трсхатомных молекулах и т.п. Такое рассмотрение во мн. случаях позволяет
предсказать наиб. вероятную равновесную геом. конфигурацию молекулы и качественно
объяснить эксперим. закономерности по изменению геом. конфигурации в рядах однотипных
молекул. При построении корреляционных диаграмм важно иметь в виду, что сохранение
симметрии молекулы при изменении ее геом. конфигурации влечет за собой сохранение
и типов симметрии отдельных орбиталей (т. наз. п р а в и л о с о х р а-н е н
и я о р б и т а л ь н о й с и м м е т р и и). При этом кривые
изменения орбитальных энергий для орбиталей одинакового типа симметрии на корреляционных
диаграммах пересекаться не могут (правило непересечения). Учет этих правил в
орг. химии позволяет предсказать, в каких р-циях энергия
активации будет велика, а в каких-мала, какой механизм активации молекул - термический
или фотохимический-более предпочтителен для данной р-ции и т.п. (см. Вудворда - Хофмана правила
).
Относит. простота учета
симметрии мол. орбиталей и определяемых этой симметрией особенностей св-в молекул
является существ. достоинством М. о. м. Именно учет симметрии мол. орбиталей
позволил ввести p-электронное приближение и для мн. сопряженных и ароматич.
систем ограничиться анализом p-электронной подсистемы в рамках простейшего из
М. о. м.-метода Хюккеля. Для расчетов спектральных св-в высокосимметричных молекул
неорг. соед. используют созданные на основе учета симметрии мол. орбиталей кристаллического поля теорию
и поля лиган-дов теорию.
Молекулярно-орбитальный
подход оказался плодотворным также для анализа электронных спектров молекул
и отнесения полос к определенным квантовым переходам. Поскольку этот подход
позволяет рассматривать отдельные орбитали, а не полную волновую ф-цию многоэлектронной
молекулы, то можно выделить именно ту часть волновой ф-ции, к-рая меняется при
электронном переходе, ионизации, в ходе хим. р-ции и т. д., и рассматривать
далее только эту часть. В частности, если предположить, что при хим. превращениях
достигается макс. перекрывание высших занятых мол. орбиталей одной молекулы
и низших вакантных мол. орбиталей другой, то при простейшем качеств. анализе
реакц. способности возможно ограничиться рассмотрением только этих орбиталей
(К. Фукуи, 1952). Условие макс. перекрывания высшей занятой мол. орбитали нафталина
и низшей свободной мол. орбитали NO2+ позволило объяснить
преим. нитрование нафталина в а-положение. В случае мономол. р-ций молекулу
условно делят на две части, одна из к-рых включает высшие занятые мол. орбитали,
а другая - низшие вакантные мол. орбитали (подробнее см. в ст. Граничных орбиталей теория
).
Подобные же соображения
используются в теории возмущений мол. орбиталей, развитой М. Дьюаром (1952).
В этой теории первоначально в я-электронном приближении рассматривалась энергия
двух реагирующих неполярных молекул на основе выражения второго порядка теории
возмущений, в к-ром осн. вклады, как правило, дают лишь слагаемые, включающие
высшие занятые и низшие свободные мол. орбитали реагирующих молекул. Простейшим
примером является взаимод. акцептора электронов А (напр., ВН3) с
донором электронов D (напр., NH3), приводящее к образованию комплекса
с переносом заряда: для такой системы осн. вклад в энергию взаимод. дает тот
член в выражении для энергии, к-рый зависит от орбитальных энергий двух орбиталей:
высшей занятой D и низшей свободной А. Разработаны и развиваются т. наз. методы
функционалов плотности, к-рые базируются на том, что по крайней мере для основного
состояния молекулы энергия есть функционал электронной плотности. В приближении
Хартри-Фока эта энергия представляет собой функционал всего лишь высшей занятой
мол. орбитали.
Локализация молекулярных
орбиталей. Долгое время считалось, что молекулярно-орбитальный подход имеет
существ. недостаток: мол. орбитали в большинстве случаев не локализованы. Электронная
плотность, отвечающая каждой орбитали, более или менее равномерно распределена
по всему объему молекулы. Классич. теория хим. строения оперирует более локальными
по своей природе образами: атомы в молекулах, хим. связи, функц. группы и т.п.
Переход от одной картины к другой легко, однако, осуществить, если в волновой
ф-ции, представляемой М. о. м. в виде определителя, выполнить такое линейное
преобразование канонич. орбиталей друг через друга (без к.-л. изменения полной
волновой ф-ции в целом), к-рое приводит к локализованным орбиталям. Эти орбитали,
как показали многочисленные расчеты, хорошо соответствуют отдельным двух-или
трехцентровым связям, неподеленным парам электронов, остовным электронам и т.
п. При таком подходе отчетливо
выделяются валентные орбитали, существенно меняющиеся при изменении геом. конфигурации
молекулы, и практически не меняющиеся остовные орбитали. При дальнейших упрощенных
подходах можно ограничиться опять-таки рассмотрением только валентных орбиталей
(работать в в а л е н т н о м п р и б л и ж е н и и). Локализованные орбитали
часто имеют практически одну и ту же форму в разл. молекулах при условии, что
локальная геом. конфигурация молекул, к к-рым относятся эти локализованные орбитали,
примерно одна и та же. Таковы, напр., локализованные орбитали С—Н связей в алканах,
локализованные орбитали функц. групп СООН, NH2 и др. На основе представлений
о локализованных орбиталях можно рассматривать закономерности, связанные с локальными
фрагментами молекул, в т.ч. с выделением функц. групп.
Наиб. существ. недостаток
М. о. м.-то, что они не учитывают электронной корреляции, т. е. взаимной согласованности
пространств. распределения электронов в многоэлектронной мол. системе. Без учета
электронной корреляции получается, что даже качеств. рассмотрение может дать
неправильные результаты для мн. возбужденных состояний молекул, в частности
при достаточно близко расположенных по энергии двух или большего числа электронных
состояний для определенных геом. конфигураций ядер. При решении подобных задач
приходится отказываться от моле-кулярно-орбитальной картины и переходить к более
сложному описанию, напр. с помощью конфигурационного взаимодействия метода
или др. неэмпирических методов
квантовой химии.
Лит.: Яцимирский
К. Б., Яцимирский В. К., Химическая связь, К., 1975; Краснов К. С., Молекулы
и химическая связь, М., 1977; Пирсон П., Правила симметрии в химических реакциях,
пер. с англ., М., 1979; Фудзина-га С., Метод молекулярных орбиталей, пер. с
япон., М., 1983.
Я. Ф. Степанов.
|


