МОНОСАХАРИДЫ
, углеводы,
представляющие собой по-лигидроксиальдегиды (альдозы) и полигидроксикетоны (ке-тозы)
общей ф-лы СnН2nОn (п = 3-9), в к-рых каждый атом С (кроме карбонильного) связан с группой ОН,
и производные этих соед., содержащие разл. др. функц. группы, а также атом Н
вместо одного или неск. гидроксилов.
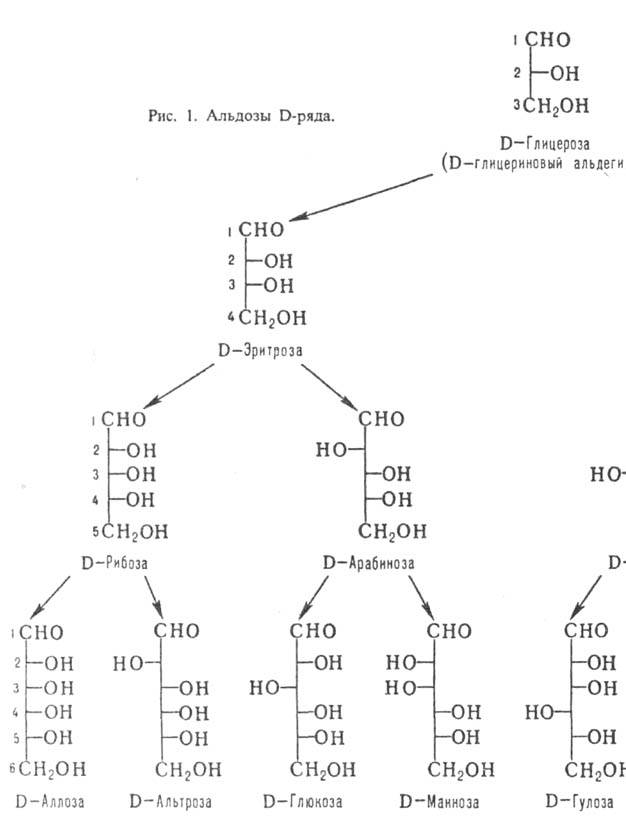
По числу атомов С различают
низшие М. (триозы и тетрозы; содержат в цепи соотв. 3 и 4 атома С), обычные
(пентозы и гексозы) и высшие (гептозы, октозы, нонозы). Углеродные атомы в молекулах
М. нумеруют таким образом, чтобы атом С карбонильной группы имел наим. номер
(рис. 1 и 2).
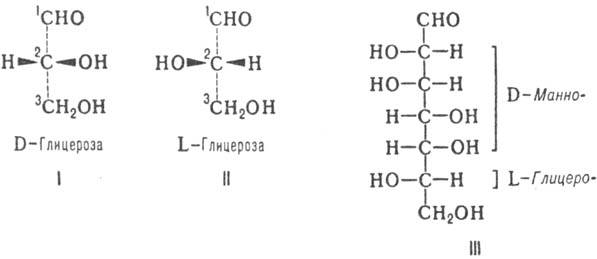
Инголда - Прелога; ф-лы
I и II). Другие М. имеют неск. асим. атомов С; их рассматривают как производные
D- или L-глицеринового альдегида и относят к D- или L-ряду (абс. конфигурация
М.) в соответствии с конфигурацией предпоследнего (п — 1) атома С.
Различия между изомерными
М. в каждом ряду обусловлены относит. конфигурацией остальных асим. центров;
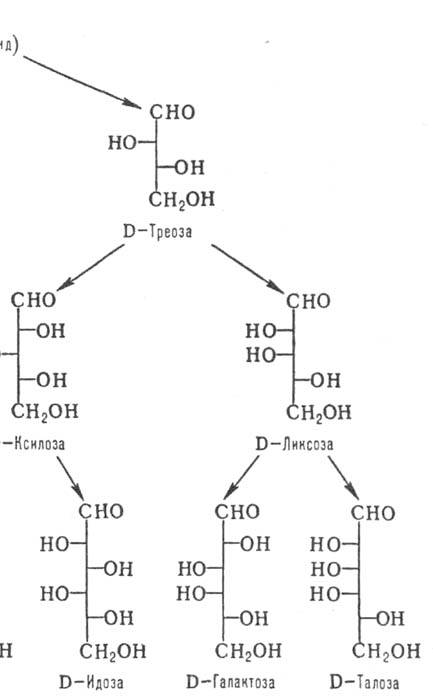
каждой относит. конфигурации в альдотетрозах, альдопен-тозах и альдогексозах
соответствует тривиальное назв. мо-носахарида (рис. 1). Для обозначения конфигураций
высших Сахаров используют префиксы, образованные из таких назв. (напр., L-глицеро-D-манно-гептоза;
III). Стереохим. взаимоотношения между М. хорошо иллюстрируются проекционными
Фишера формулами
, в к-рых группа ОН располагается справа от вертикальной
черты, обозначающей углеродную цепь, если соответствующий асим. центр имеет
D-конфигу-рацию, и слева, если он имеет L-конфигурацию. Каждому представителю
D-ряда соответствует его оптич. антипод, относящийся к L-ряду, в к-ром все асим.
центры имеют противоположную конфигурацию. Общее число изомерных альдоз равно
2n, где п-число асим. атомов С в молекуле.
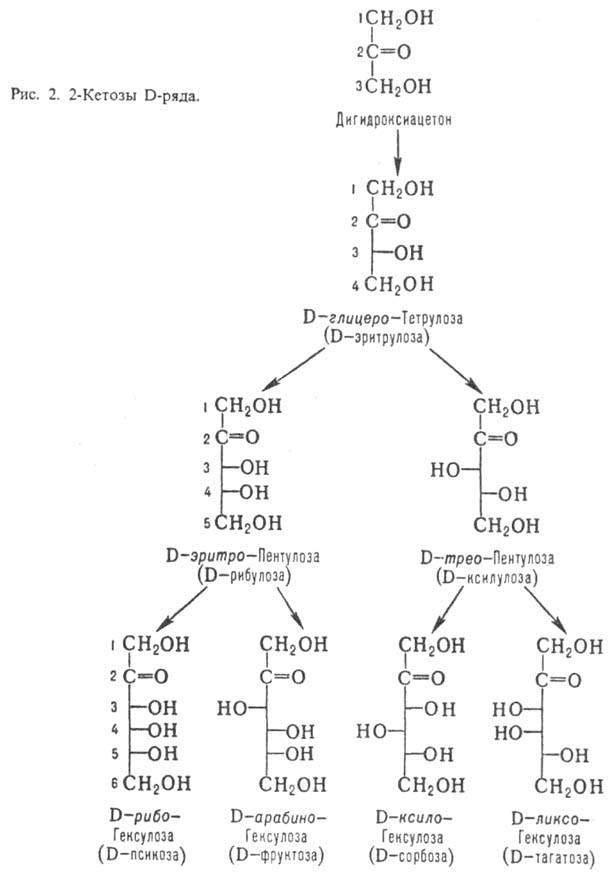
Кетозы по сравнению с альдозами
с той же длиной углеродной цепи содержат на один асим. атом С меньше. Однако
у них встречается еще один вид изомерии, обусловленный разл. положением карбонильной
группы. У большинства прир. кетоз атом С карбонильной группы занимает положение
2; представители D-ряда таких кетоз показаны на рис. 2.
Карбонильные группы М.
легко реагируют внутримолеку-лярно с группами ОН, образуя циклич. полуацетали.
Последние могут представлять собой пятичленный (фураноз-ная форма) или шестичленный
(пиранозная форма) цикл и отличаться конфигурацией образующегося на месте карбонильной
группы нового асим. центра (атом С этого асим. центра наз. а н о м е р н ы м,
или г л и к о з и д н ы м). Эту конфигурацию обозначают буквой а, если она совпадает
с конфигурацией центра, определяющего принадлежность М. к D- или L-ряду, и буквой
Р в противоположном случае. Изомеры, различающиеся лишь конфигурацией аномерного
атома, наз. а н о м е р а м и, а изменение конфигурации при этом атоме-а н о
м е р и з а ц и е й. Для изображения циклич. форм М. удобно пользоваться Хоуорса формулами
(см., напр., ф-лы IV и V-пиранозная и фуранозная формы кето-зы,
а также рис. 3-циклич. ф-лы альдозы).

В р-ре каждый М. находится
в виде смеси таутомеров (напр., рис. 3), соотношение между к-рыми в состоянии
равновесия определяется их термодинамич. устойчивостью; в большинстве случаев
преобладают пиранозные формы, а ациклические
присутствуют в следовых кол-вах. Напротив, М. в кристаллич. состоянии представлены
одной из тауто-мерных форм. Растворение кристаллов сопровождается таутомерными
превращ., за протеканием к-рых можно следить по изменению во времени величины
оптич. вращения (это явление наз. мутаротацией
).

Рис. 3. Таутомерное
равновесие D-глюкозы.
Фуранозные формы М. термодинамически
менее выгодны, чем пиранозные, поскольку в практически плоском пятичленном цикле
заместители вынуждены находиться в нестабильной заслоненной конформации. Напротив,
шести-членные циклы имеют кресловидную форму, в к-рой заместители при соседних
атомах С занимают более выгодные положения, соответствующие скошенной конформации.
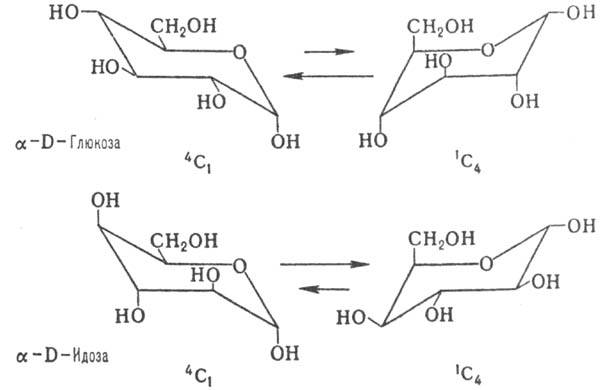
Существуют два типа кресловидной конформации пираноз-1С4
и 4C1 (цифры в верх. и ниж. индексах показывают номера
атомов С, находящихся соотв. в верх. и ниж. положениях кресла). Более устойчивым
является кресло с наим. числом объемистых аксиальных группировок. Поэтому для
большинства альдогексоз D-ряда предпочтительным является кресло 4C1
с экваториальной группой СН2ОН и только для D-идозы в равновесии
преобладает конформа-ция 1С4 (влияние накопления аксильных
групп ОН):
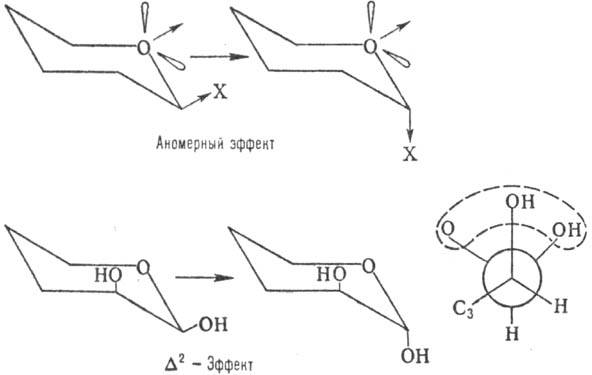
На относит. устойчивость
a- и b-форм кроме пространств. факторов влияют диполь-дипольные взаимод.-аномерный
эффект, в результате к-рого заместитель при аномерном атоме стремится занять
аксиальное положение (особенно в малополярных р-рителях), и D2-эффект,
заключающийся в повыш. нестабильности конформера с экваториальным заместителем
у аномерного атома при наличии аксиального гидроксила в положении 2 (вследствие
взаимного отталкивания атомов О; см. ф-лу Ньюмена):
Кроме того, на относит.
устойчивость a- и b-форм влияют водородные связи, к-рые могут играть стабилизирующую
роль в относительно малополярных р-рителях, и нек-рые др. факторы.
Кроме обычных известно
неск. групп М., отличающихся своеобразным набором функц. групп или структурой
углеродной цепи. К ним относятся дезоксисахара
(одна или неск. групп
ОН замещены на атомы Н), аминосахара
(одна или неск. групп ОН замещены
на аминогруппы), урановые кислоты (группа СН2ОН окислена в
карбоксильную), разветвленные сахара (имеют разветвленную углеродную цепь с
ме-тильной, гидроксиметильной или альдегидной группой в качестве ответвлений),
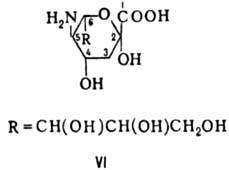
высшие 2-кето-З-дезоксиальдоновые к-ты, в т.ч. сиаловые к-ты - производные 2-кето-3,5-дидез-окси-5-амино-D-глицеро-D-галакто-нононовой (нейрамино-вой) к-ты (ф-ла VI).
Физические и химические
свойства. М.-бесцв. кристаллич. в-ва, легко раств. в воде, ДМСО, трудно-в
этаноле, не раств. в неполярных орг. р-рителях. Важнейшие физ.-хим. характеристики
М.-уд. вращение, используемое наряду с хроматографич. методами для идентификации
природных М., и спектры ПМР, с помощью к-рых можно определить относит. конфигурацию
М. Для выяснения абс. конфигурации используют рентгеноструктурный анализ кристаллов
или хим. трансформацию М. в более простые соед. с известной конфигурацией.
Хим. св-ва М. обусловлены
наличием в их молекулах групп С=О (у ациклич. таутомера) и ОН. При действии
NaBH4 в водном р-ре карбонильная группа М. количественно восстанавливается
до спиртовой; образующиеся полиолы в виде летучих ацетатов или триметилсилиловых
эфиров можно использовать для количеств. анализа смесей М. с помощью ГЖХ. Группы
С=О альдоз в мягких условиях окисляются бромной водой с образованием лактонов
альдо-новых к-т. Кетозы в эту р-цию не вступают и м. б. таким образом выделены
из сложных смесей с альдозами. Определение "восстанавливающей способности"
(т.е. окисление группы С=О) используют в многочисл. методиках анализа М.
При действии оснований
возможна енолизация группы С=О, сопровождающаяся изменением конфигурации соседнего
асим. центра или миграцией карбонила (см. Лобри де Брюйна - ван Экенстейна реакция
). В более жестких условиях
происходят b-элиминирование заместителей и скелетные перегруппировки. При обработке
к-тами в жестких условиях из пентоз образуется фурфурол, из гексоз - 5-гид-роксиметилфурфурол;
конденсация последних с фенолами или ароматич. аминами с образованием окрашенных
соед. лежит в основе разнообразных методик спектрофотометрич. определения М.
Из р-ций группы С=О с азотистыми
соед. большое историч. значение имела конденсация М. с фенилгидрази-ном, приводящая
к фенилозазонам. Широко используется р-ция альдоз с NH2OH с послед.
ацетилированием:
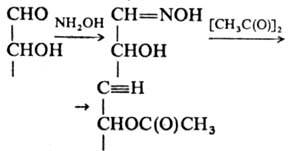
Образующиеся ацетилир.
альдононитрилы удобны для анализа с использованием ГЖХ. Распад таких нитрилов
под действием оснований служит для укорочения углеродной цепи М. на один атом,
а присоединение HCN к карбонильной группе с послед. восстановлением нитрила
в альдегид (Килиани - Фишера реакция) - для удлинения.
Из р-ций групп ОН широко
используются образование простых и сложных эфиров, ацеталей, замещение на др.
функцией, группировки, окисление и т. д. Метиловые эфиры (весьма прочные соед.)
служат для постоянной защиты групп ОН в структурном анализе олиго- и полисахаридов.
Бензиловые эфиры применяют для временной защиты групп ОН, поскольку эти группы
легко удаляются гидрогено-лизом. Трифенилметиловые (тритиловые) эфиры удобны
для избират. замещения первичных групп ОН в присут. вторичных. Ацетилирование
широко применяют для временной неизбират. защиты всех групп ОН в молекуле М.,
тогда как бензоилирование чаще используют для избират. замещения (первичные
группы ОН реагируют легче вторичных, экваториальные легче аксильных). Омыление
эфиров, образованных орг. к-тами, легко осуществляется обработкой метилатом
Na в метаноле и приводит к исходным М. Напротив, эфиры сульфокислот (тозилаты,
мезилаты и трифлаты) применяют для активации соответствующей группы ОН при нуклеоф.
замещении, в т.ч. с обращением конфигурации. Это позволяет осуществлять эпимеризацию
(напр., при действии бензоата Na) у к.-л. атома С (т.е. переход от одного М.
к другому), замещение группы ОН на атом галогена, на др. функц. группы (синтез
амино- и тиосахаров), получать внутримол. простые эфиры (ангид-росахара), ненасыщенные
производные М. и т.д.
С альдегидами и кетонами
М. образуют циклич. ацетали, строение к-рых определяется природой исходного
М. и реагента (с альдегидами предпочтительно образуются шес-тичленные циклы,
с кетонами -пятичленные); р-ция может сопровождаться таутомерными превращ. (знак
~ показывает, что М. может иметь a- или b-конфигурацию), напр.:
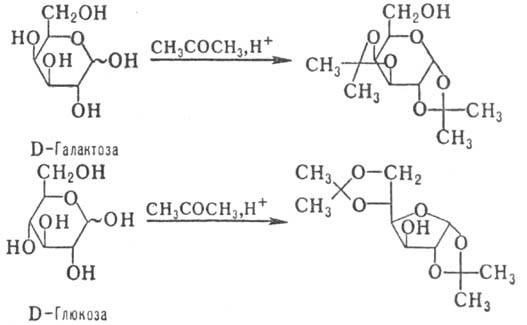
Ацетали устойчивы в щелочных
и нейтральных средах; в присут. к-т гидролизуются, причем в ряде случаев возможно
избират. удаление одной из двух присутствующих группировок, напр.:
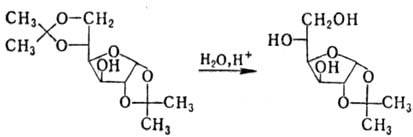
Комбинации перечисл. р-ций
групп ОН (образование и гидролиз простых и сложных эфиров, получение ацеталей)
позволяют синтезировать частично защищенные М. с определенным расположением
заместителей, что имеет важное значение в синтетич. химии М.
Окисление защищенных М.,
содержащих своб. первичную группу ОН, применяют для получения уроновых к-т,
а содержащих своб. вторичную группу ОН-для получения карбонильных производных,
широко используемых как про-межут. соед. в синтезе М. с новыми функц. группами,
для разветвления углеродной цепи и для обращения конфигурации асим. центра при
послед. восстановлении. Для анализа М. применяют специфич. окисление их йодной
к-той и ее солями-т. наз. периодатное окисление (см. Малапрада реакция
).
Гидроксил у аномерного
атома С (его называют аномер-ным, гликозидным, или полуацетальным) значительно
отличается от прочих групп ОН повыш. склонностью к р-циям нуклеоф. замещения.
Его обмен на остатки спиртов приводит к образованию гликозидов
. Если
спиртовой компонентой гликозида (агликоном) служит др. молекула М., образуются
олигосахариды
и полисахариды
. При кипячении М. с большим избытком
низшего спирта в присут. к-т (метод Фишера) образуется смесь изомерных гликозидов,
соответствующих разным таутомерным формам М.:
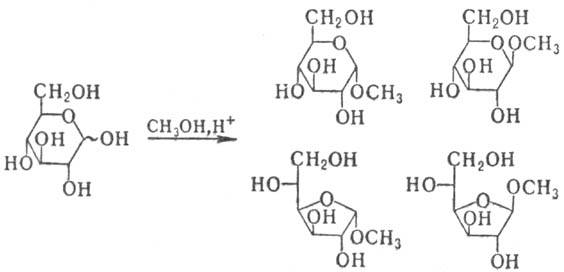
При разделении этой смеси
получают индивидуальные гли-козиды, устойчивые в щелочных и нейтральных средах,
а при нагр. с водными к-тами дающие исходный М. Получение таких гликозидов служит
наиб. удобным и распространенным способом временной защиты карбонильной группы
М.
Для синтеза гликозидов
со сложными агликонами применяют защищенные производные М. с фиксир. размером
цикла, в к-рых гликозидный центр активирован введением подходящего заместителя
(см., напр., Кёнигса-Кноppa
[Кенигса-Кноppa] реакция
).
Распространение в природе.
В природе широко распространены гексозы (D-глюкоза, D-галактоза, D-манноза,
D-фруктоза) и пентозы (D-ксилоза, D-арабиноза, D-рибоза). М. редко в природе
встречаются в своб. состоянии. Они входят в виде остатков в состав многочисл.
гликозидов: олиго- и полисахаридов, более сложных биополимеров-гли-копротеинов,
гликолипидов
, нуклеиновых кислот
и др. Исключение - D-глюкоза, содержащаяся
в плазме крови и соках
растений, и D-фруктоза, большие кол-ва к-рой имеются в меде и плодах нек-рых
растений.
Образование М. в растениях
связано с ассимиляцией ими СО2 и происходит в результате фотосинтеза
.
Молекула СО2 присоединяется к 1,5-дифосфату D-рибулозы в хлоро-пластах
с участием фермента рибулозодифосфат-карбокси-лазы, а образующаяся в результате
3-фосфо-D-глицериновая к-та (ф-ла VII) путем дальнейшего восстановления и конденсаций
дает D-глюкозу (см. Глюконеогенeз
)или D-фруктозу; при этом регенерируется
молекула рибулозодифосфата (цикл Кальвина):
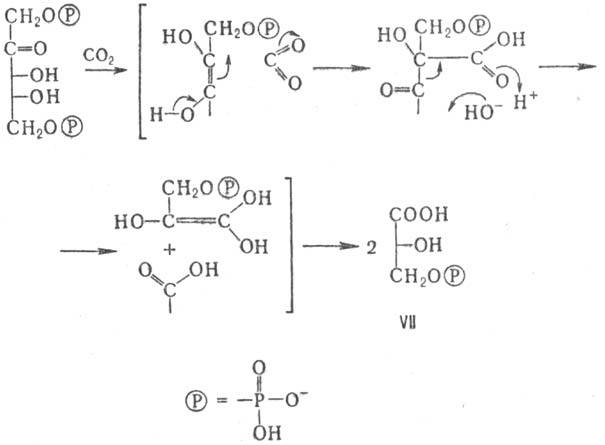
Распад гексоз, окислительный
или анаэробный (гликолиз
), служит источником энергии для большинства
растит. и животных клеток; ферментативные трансформации гексоз приводят к получению
др. природных М., а при более глубоких превращ. в обмене веществ
из них
образуются аминокислоты, липиды и др. орг. соединения. В многочисл. р-ции окисления,
гликозилирования, восстановления и эпи-меризации под действием соответствующих
ферментов М. вступают в виде нуклеозиддифосфатсахаров (НДФС). Так, напр., из
уридиндифосфат-D-глюкозы (VIII) могут образовываться уридиндифосфат-D-галактоза
(IX) и уридиндифос-фат-D-глюкуроновая к-та (X):
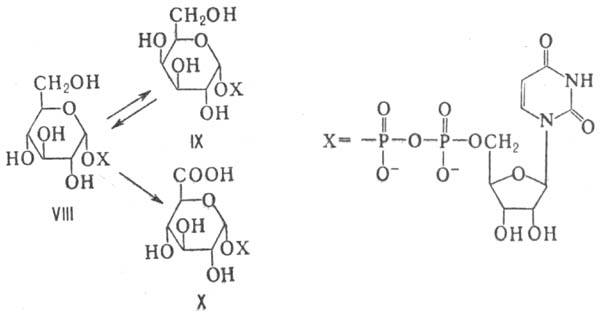
НДФС служат в живых клетках
для построения гли-козидной связи; причем в зависимости от природы фермента,
катализирующего гликозилирование, эта р-ция может сопровождаться как сохранением,
так и обращением конфигурации аномерного атома С.
В биосинтезе углеводсодержащих
биополимеров с участием мембранных ферментов в качестве доноров гликозиль-ных
остатков выступают др. активированные производные М., наделенные липофильными
св-вами-полипренилмоно-или полилренолдифосфатсахара; напр., бактопренол-11-ди-фосфатглюкоза
(XI)
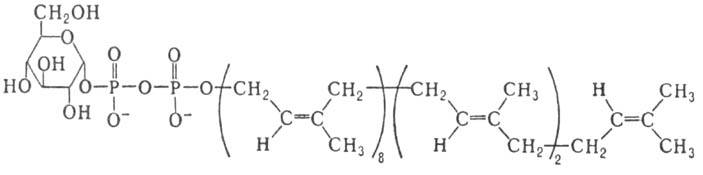 XI
XI
Получение и применение.
М. получают кислотным гидролизом полисахаридов (напр., D-глюкозу-из крахмала,
D-ксилозу-из богатых ксиланами отходов переработки с.-х. растений и древесины).
Смесь глюкозы с фруктозой получают гидролизом сахарозы и используют в пищ. промети.
D-Глюкоза находит применение в медицине. Восстановление D-глюкозы в D-сорбит
и D-ксилозы в ксилит осуществляют в пром. масштабах водородом над никелевым
катализатором. D-Сорбит служит исходным соед. в синтезе аскорбиновой к-ты (см. Витамин С
)и наряду с ксилитом используется как обладающий сладким вкусом
заменитель сахарозы при заболевании диабетом. Разнообразные М. часто служат
удобными хиральными исходными в-вами в синтезе сложных прир. соед. неуглеводной
природы.
Лит.: Химия углеводов,
пер. с англ., М., 1967; Стоддарт Дж., Стереохимия углеводов, пер. с англ., М.,
1975; Степаненко Б. Н., Химия и биохимия углеводов. Моносахариды, М., 1977;
Общая органическая химия, пер. с англ., т. 11, М., 1986, с. 127-202; Khadem
Hassan S. El., Carbohydrate chemistry. Monosaccharides and their oligomers,
San Diego, 1988. А. И. Усов.
|


