РАДИКАЛЬНАЯ ПОЛИМЕРИЗАЦИЯ
,
полимеризация
, в к-рой активные центры роста представляют собой своб.
радикалы. Р. п. возможна для большинства виниловых, винилиденовых, диеновых
мономеров, а также для нек-рых напряженных циклич. соединений. Нек-рые ненасыщ.
мономеры не способны полимеризоваться по радикальному механизму вследствие стерич.
затруднений (напр., 1,2-дизаме-щенные виниловые мономеры) или в случае вырожденной
передачи цепи (см. ниже), напр. пропилен, аллильные мономеры.
Р. п.-один из осн. пром.
методов, к-рым получают более половины производимых в мире полимеров, в т. ч.
полиэтилен
(высокого давления), полистирол
, сополимеры этилена
и стирола с разл. полярными мономерами, поливинилхлорид
, полиакрилаты
и
полиметакрилаты
, ряд синтетич. каучуков и водорастворимых карбоцепных
полиэлектролитов.
Кинетич. схема Р. п. включает
четыре элементарные стадии: инициирование, рост, передачу и обрыв цепи. На стадии
инициирования образуются первичные радикалы мономера в результате непосредств.
энергетич. воздействия (тепло, УФ либо ионизирующее излучение; о двух последних
см. Фотополимеризация
, Радиационная полимеризация
)или чаще при взаимод.
мономера с радикалами, возникающими при гомолитич. распаде специально вводимых
в-в-инициаторов радикальных
(напр., пероксидов, гидропероксидов, азосоединений).
Для увеличения скорости инициирования при низких т-рах к пероксидам добавляют
восстановители, напр. соли переходных металлов или амины (т. наз. окис-лит.-восстановит.
инициаторы).
Стадия инициирования включает
по меньшей мере два последоват. элементарных акта - генерирование радикалов
R• (р-ция 1, а) и их взаимод. с мономером (р-ция 1, б):


(I-инициатор, М-мономер,
М•1-первичный мономерный радикал, k1
и k1 - константы скоростей соответствующих р-ций).
Помимо р-ций (1, б), радикалы R• могут участвовать в побочных р-циях,
что учитывают с помощью коэф. эффективности инициирования (f); последний
характеризует отношение числа радикалов, участвующих в р-ции (1, б), к общему
числу радикалов, образовавшихся по р-ции (1, а). Значения f обычно составляют
0,6-0,8, a k1 и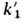 имеют порядок 10-5-10-4 с-1 и 10-103
л/(моль·с) соотв., поэтому
имеют порядок 10-5-10-4 с-1 и 10-103
л/(моль·с) соотв., поэтому 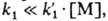 где [М]-концентрация мономера (в моль/л) в реакц. среде; таким образом акт (1,
а) кинетически определяет стадию инициирования:
где [М]-концентрация мономера (в моль/л) в реакц. среде; таким образом акт (1,
а) кинетически определяет стадию инициирования:

где u1-скорость
инициирования.
Осн. стадия полимеризации-р-ция
роста цепи, при многократном повторении к-рой образуется макромол. цепь, - описывается
ур-нием:

(M•n-макрорадикал,
содержащий п мономерных звеньев). Скорость р-ции роста выражается ур-нием:

При этом предполагается,
что реакц. способность макрорадикалов не зависит от их длины; как правило, такое
предположение справедливо при п > 3-5. Кинетич. параметры р-ции (3)
приведены в табл. 1.
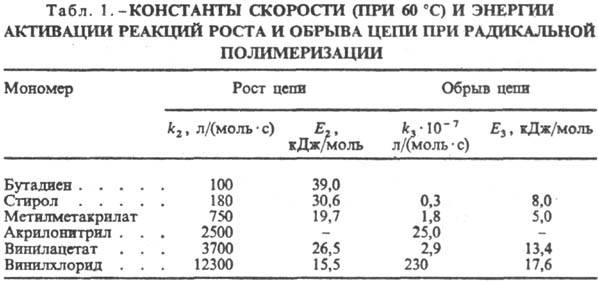
Значения k2
зависят от двух факторов - реакц. способностей (активностей) мономера и
макрорадикала, к-рые, как правило, изменяются в противоположных направлениях,
поскольку реакц. способность мономера при наличии в его молекуле сопряженных
связей повышается, а макрорадикала снижается. Из этих двух факторов на скорость
роста цепи определяющее влияние оказывает активность макрорадикала, поэтому
последовательность расположения мономеров в табл. 1 отражает рост реакц. способности
макрорадикалов. Реакц. способности мономеров и радикалов зависят также от полярного
и стерич. факторов, наиб. полно проявляющихся в р-циях сополимеризации (см. Сополимеры
)или др. конкурирующих р-циях. Скорости и направление радикальных
р-ций обычно мало зависят от характеристик реакц. среды. Однако при наличии
специфич. взаимодействий мономера и (или) радикала с молекулами среды, напр.
при образовании p-комплексов, донорно-акцепторных комплексов, комплексов
с участием к-т Льюиса или водородных связей, наблюдается изменение констант
скорости роста цепи.
Присоединение мономеров
при Р. п. происходит преим. по типу "голова к хвосту":
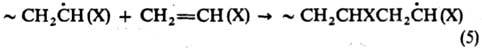
Доля звеньев, присоединенных
по типу "голова к голове" и "хвост к хвосту" обычно
не превышает неск. процентов и уменьшается при полимеризации мономеров, содержащих
объемные заместители X. В то же время не происходит стереорегулярного присоединения.
Так, при Р. п. виниловых мономеров образуются атактич. полимеры с нек-рым преобладанием
синдиотактич. структур (см. Стереорегулярные полимеры
). Снижение
т-ры полимеризации способствует увеличению доли синдиотактич. структур.
Ограничение растущих цепей
при Р. п. возможно путем обрыва и (или) передачи цепи. Обрыв цепи-необратимая
хим. дезактивация растущих цепей-протекает обычно в результате диспропорционирования
двух макрорадикалов (р-ция 6, а) или их рекомбинации (6, б):
.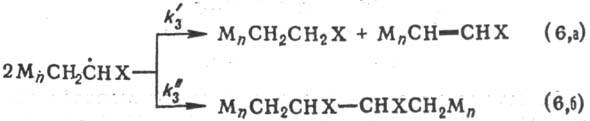
(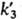 и
и
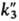 -константы скоростей
соответствующих р-ций). Вклад р-ций (6, а) или (6, б) в общую скорость обрыва
определяется активностью взаимодействующих макрорадикалов и стерич. факторами:
для малоактивных радикалов и при наличии небольших заместителей X в мономере,
как правило, характерен обрыв по механизму рекомбинации. Скорость р-ции обрыва
цепи выражается ур-нием:
-константы скоростей
соответствующих р-ций). Вклад р-ций (6, а) или (6, б) в общую скорость обрыва
определяется активностью взаимодействующих макрорадикалов и стерич. факторами:
для малоактивных радикалов и при наличии небольших заместителей X в мономере,
как правило, характерен обрыв по механизму рекомбинации. Скорость р-ции обрыва
цепи выражается ур-нием:

Здесь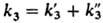 -суммарная
константа скорости обрыва, к-рая
на начальных стадиях превращения имеет порядок 106-107
л/(моль·с) (см. табл. 1).
-суммарная
константа скорости обрыва, к-рая
на начальных стадиях превращения имеет порядок 106-107
л/(моль·с) (см. табл. 1).
Бимолекулярный обрыв цепи-диффузионно-контролиру-емая
р-ция, состоящая из трех последоват. стадий: 1) посту-пат, диффузия двух макрорадикалов
с образованием объединенного клубка; 2) взаимное сближение активных концов вследствие
диффузии отдельных звеньев и сегментов цепи; 3) непосредств. хим. взаимодействие
реакц. центров с образованием неактивных макромолекул. Для большинства изученных
виниловых мономеров k3 обратно пропорциональна вязкости исходной
системы, а скорость обрыва лимитируется стадией 2. Так, факторы, снижающие подвижность
сегментов цепи (введение сомономера, увеличивающего жесткость цепи, снижение
т-ры полимеризации и др.), значительно уменьшают скорость бимолекулярного обрыва.
Передача цепи -р-ция, приводящая
к переносу активного центра от растущего макрорадикала на любую др. молекулу
(р-ритель, мономер, инициатор, полимер), выступающую в роли агента передачи
(А), с образованием "мертвой" макромолекулы (Мn)
и нового активного центра (А•):
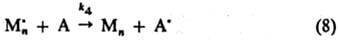
(k4
- константа скорости передачи цепи). Обычно р-ция передачи цепи приводит
к продолжению кинетич. цепи, поскольку новый радикал А• способен
инициировать Р. п. со скоростью, близкой к скорости р-ции роста. В противном
случае имеет место вырожденная передача цепи (т.е. рост цепи происходит с меньшей
скоростью на А•, чем на макрорадикале) либо ингибирование Р. п. (см. Ингибиторы
).
Осн. р-ции передачи цепи-перенос
подвижного атома Н или Hal от агента передачи к макрорадикалу, реже-в обратном
направлении. Скорость р-ции передачи цепи и место отщепления атома Н или Hal
определяются энергией разрывающейся в молекуле агента передачи связи С—Н, S—Н,
С—Hal и т.п. Так, скорость отщепления атомов Н в насыщ. углеводородах уменьшается
в ряду: водород при третичном атоме С > при вторичном > первичном.
Р-ции передачи цепи конкурируют
С р-цией роста, поэтому для количеств. характеристики передачи обычно используют
константы передачи С = k4/k2, по
к-рым судят о реакц. способности агентов передачи.
Внутримол. и межмол. р-ции
передачи цепи на полимер приводят к образованию макромолекул с короткоцепными
разветвлениями, сшитых или привитых сополимеров. На практике р-ции передачи
цепи используют для регулирования мол. массы полимера и для синтеза привитых сополимеров
(путем передачи цепи на полимер). В первом случае используют
агенты передачи с С > 10-3 , к-рые наз. регуляторами мол.
массы (см. табл. 2). При С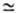 1
в ходе р-ции образуются низкомол. продукты (см. Теломеризация
).
1
в ходе р-ции образуются низкомол. продукты (см. Теломеризация
).
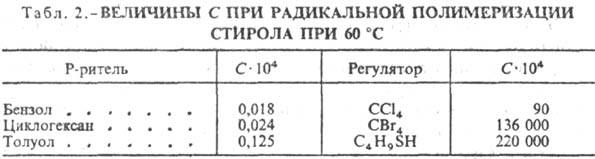
На начальных стадиях превращения
Р. п., как правило, протекает с постоянной скоростью, что связано с выполнением
условия квазистационарности (постоянства концентрации активных центров):

Последнее позволяет определить
концентрацию активных центров, к-рую находят подстановкой значений u1
и u3 из ур-ний (2) и (7) в ур-ние (9):
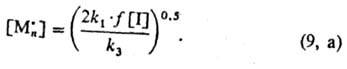
Поскольку мономер расходуется
гл. обр. на стадии роста цепи, общая скорость v P. п. практически равна
скорости этой стадии
и м. б. определена подстановкой выражения (9, а) в ур-ние (4):
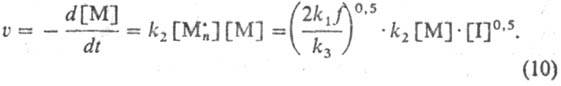
Первый порядок по мономеру
и половинный по инициатору наиб. типичны для Р. п. Др. важнейший кинетич. параметр
Р. п.-средняя мол. масса, или средняя степень полимеризации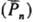 ,
образующегося полимера определяется отношением скорости роста цепи к сумме скоростей
обрыва и передачи цепи:
,
образующегося полимера определяется отношением скорости роста цепи к сумме скоростей
обрыва и передачи цепи:

В условиях квазистационарности
и обрыва цепи диспропор-ционированием справедливо ур-ние:
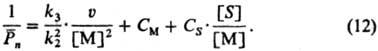
Здесь См
и Cs- константы передачи цепи на мономер и р-ритель (S).
В случае обрыва цепи рекомбинацией радикалов в первое слагаемое вводится
множитель 1/2.
Величина См
определяет верх. возможный предел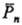 .
Для виниловых мономеров См
.
Для виниловых мономеров См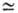 10-4-10-5,
поэтому из них можно получать высокомол. полимеры. В то же время- из-за высоких
значений См мономеров аллилового ряда (напр., для аллилацетата
См
10-4-10-5,
поэтому из них можно получать высокомол. полимеры. В то же время- из-за высоких
значений См мономеров аллилового ряда (напр., для аллилацетата
См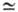 0,1),
пропилена и др. a-олефинов, обусловленных высокой подвижностью атомов Н
в a-поло-жении к связи С=С, получить из таких мономеров полимеры обычными
методами не удается.
0,1),
пропилена и др. a-олефинов, обусловленных высокой подвижностью атомов Н
в a-поло-жении к связи С=С, получить из таких мономеров полимеры обычными
методами не удается.
Полимерам, образующимся
при Р. п., свойственно широкое молекулярно-массовое распределение
(ММР),
детальный характер к-рого в условиях гомог. процесса определяется механизмом
ограничения растущих цепей. Так, при ограничении растущих цепей по р-циям диспропорционирования
и (или) передачи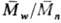 =2
(где
=2
(где 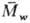 и
и 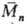 -соотв.
среднемассовая и среднечисловая
мол. массы), а при обрыве рекомбинацией
-соотв.
среднемассовая и среднечисловая
мол. массы), а при обрыве рекомбинацией 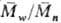 =
1,5. При параллельном осуществлении
обеих р-ций бимолекулярного обрыва это отношение имеет промежут. значение. При
Р. п. до глубоких степеней превращения мономеров или при образовании нерастворимого
полимера наблюдается значит. уширение ММР вплоть до появления полимодального
ММР.
=
1,5. При параллельном осуществлении
обеих р-ций бимолекулярного обрыва это отношение имеет промежут. значение. При
Р. п. до глубоких степеней превращения мономеров или при образовании нерастворимого
полимера наблюдается значит. уширение ММР вплоть до появления полимодального
ММР.
С увеличением степени превращения
мономера в ходе Р. п. происходят существ. изменения состава и физ. св-в реакц.
среды, к-рые отражаются на кинетике р-ции и характеристиках образующихся продуктов.
Так, значит. увеличение вязкости реакц. среды ограничивает в первую очередь
диффузионную подвижность макрорадикалов и, следовательно, снижает скорость обрыва,
приводя к увеличению скорости Р. п. и мол. массы образующегося полимера (гель-эффект).
При образовании нерастворимого полимера подобные явления проявляются уже в начале
процесса вследствие иммобилизации ("застревания") растущих цепей
в матрице полимера.
В Р. п. широко используют
полифункцион. инициаторы, мономеры, агенты передачи цепи, повторное участие
к-рых в ходе полимеризации изменяет структуру полимера или кинетич. характеристики.
Так, полиинициаторы способны придавать Р. п. кинетич. закономерности поликонденсации,
из полифункцион. мономеров образуются сшитые полимеры, а введение полифункцион.
агентов передачи цепи приводит к получению звездообразных полимеров.
Р. п. может быть осуществлена
в массе, эмульсии, суспензии, р-рс и др. средах (см., напр., Блочная полимеризация
,
Полимеризация в растворе
, Эмульсионная полимеризация
, Суспензионная полимеризация
,
Газофазная полимеризация
).
Первые наблюдения о возможности
самопроизвольной (термической) полимеризации высокоактивных мономеров (стирол,
винилхлорид) сделаны еще в сер. 19 в. Однако представление
о Р. п. как о радикально-цепном процессе, состоящем из совокупности элементарных
актов, начали складываться в 30-40-е гг. 20 в. (Г. Штаудингср. П. Флори,
С, С. Медведев, C.B. Лебедев) в тесной связи с развитием общих представлений
о цепных р-циях и своб. радикалах (см. Цепные реакции
, Радикалы свободные
).
Лит.: Багдасарьян
Х.С., Теория радикальной полимеризации, М., 1966; Оудиан Дж., Основы химии полимеров,
пер. с англ., М., 1974; Глады-шсв Г. П., Попов В. А., Радикальная полимеризация
при глубоких степенях превращения, М., 1974: Иванчев С. С., Радикальная полимеризация,
Л., 1985; Кабанов В. А., Зубов В. П., Семчиков Ю.Д., Комплексно-радикальная
полимеризация, М., 1987. М. Б. Лачинов. В. П. Зубов.
|


