АМИНЫ
[от ам (миак)], производные NH3,
атомы Н в к-рых замещены на орг. остатки R. В зависимости от числа R А.
делят на первичные RNH2, вторичные RRNH и третичные RRR"N;
R,R,R" м.б. одинаковыми или разными. Соотв.—NH2 наз. первичной
аминогруппой,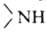 -вторичной и
-вторичной и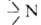 -третичной.
По числу аминогрупп в молекуле различают моно-, ди-, три- и полиамины.
-третичной.
По числу аминогрупп в молекуле различают моно-, ди-, три- и полиамины.
К последним относится, напр., тетраэтиленпентамин H2N(CH2CH2NH)3CH2CH2NH2.
Известны также циклич. амины, напр. пиперидин, хинуклидин.
Названия А. образуют от названий орг. остатков, связанных с атомом N,
напр. CH3NH2-метиламин, СН3NHС3Н7
-
метилпропиламин, (С2Н5)3N - триэтиламин.
Используются также названия, образованные прибавлением приставки "амино",
"диамино" и т.д. к обозначению родового углеводорода, например соединение
типа С2Н5СН(NН2)СН2СН3
-
3-аминопентан. Многие ароматич. А. имеют тривиальные названия, напр. C6H5NH2
- анилин, СН3С6Н4NН2
-
толуидины и СН3ОС6Н4NН2 - анизидины
(соотв. от "толуол" и от "анизол"). Высшие алифатич. А. нормального строения
иногда наз. по наименованиям радикалов жирных к-т, из к-рых А. были синтезированы,
напр. стеариламин, трилауриламин.
В ИК-спектрах характерные валентные колебания связей NH в р-ре наблюдаются
для первичных алкиламинов в областях 3380-3400 см-1 и
3320-3340 см-1; для первичных ароматич. А. - две полосы
поглощения в области 3500-3300 см-1 (обусловлены симметричными
и несимметричными валентными колебаниями связей N—Н); для алифатич. и ароматич.
вторичных А.-одна полоса соотв. в области 3360-3310 см-1 и
в области 3500-3300 см-1; третичные А. в этой области
не поглощают. В спектрах ЯМР хим. сдвиг протона аминогруппы составляет
1-5 м.д. Алифатич. А. в УФ и видимой областях не поглощают, ароматич. А.
в УФ-спектрах имеют две полосы поглощения, обусловленные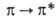 -переходами.
-переходами.
Алкиламины-сильные основания, ариламины менее ос-новны. При взаимод.
с минеральными к-тами А. образуют соли, в большинстве случаев р-римые в
воде: RNH2 + НС1 -> [R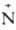 Н3]Сl-.
При реакции, напр., первичных А. с алкилгалогенидами образуются смеси А.
различной степени алкилирования, а также четвертичные соли аммония.
Н3]Сl-.
При реакции, напр., первичных А. с алкилгалогенидами образуются смеси А.
различной степени алкилирования, а также четвертичные соли аммония.
При нагр. с карбоновыми к-тами, их ангидридами, хлор-ангидридами или
сложными эфирами первичные и вторичные А. ацилируются с образованием N-замещенных
амидов, напр.: RNH2 + СН3СООН -> RNHCOCH3
+ Н2О. Ангидриды реагируют в мягких условиях, еще легче - хлорангидриды,
ацилирование к-рыми проводят в присут. основания, связывающего образующийся
в р-ции НС1. При поликонденсации диаминов с дикарбоновыми к-тами, их эфирами
или хлорангидридами образуются полиамиды. Ацилированные А. обладают слабыми
основными св-вами.
Под действием HNO2 алифатич. первичные А. превращаются в
спирты с выделением N2 и Н2О, вторичные - в N-нитрозамины
R2NNO. Третичные А. при обычной т-ре с HNO2 не реагируют.
Р-ция с HNO2 применяется для идентификации алифатич. А. При
взаимод. первичных ароматич. А. с HNO2 в кислой среде образуются
соли диазония: ArNH2 + HNO2 + НС1 -> Аr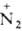 Сl-
+ 2H2O. В тех же условиях вторичные ароматич. А. превращаются
в N-нитрозамины, третичные - в пара
-нитрозопроизводные. Первичные
алициклич. А. с HNO2 образуют спирты, что часто сопровождается
сужением или расширением цикла (см. Демьянова перегруппировка
).
Сl-
+ 2H2O. В тех же условиях вторичные ароматич. А. превращаются
в N-нитрозамины, третичные - в пара
-нитрозопроизводные. Первичные
алициклич. А. с HNO2 образуют спирты, что часто сопровождается
сужением или расширением цикла (см. Демьянова перегруппировка
).
Алифатич. первичные и вторичные А. взаимод. с С12 или Вr2,
образуя N-галогензамещенные. Первичные А. с фосгеном СОС12 образуют
изоцианаты RNCO или дизамещенные мочевины (RNH)2CO, вторичные
А. - тетразамещенные мочевины R2NCONR2. Первичные
А. легко взаимод. с альдегидами, давая азометины (основания Шиффа), напр.:
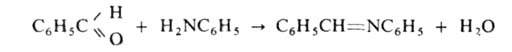
При взаимодействии первичных и вторичных А. с этиленхлоргидрином образуются
гидроксиэтильные производные, например: C6H5NH2+ С1СН2СН2ОН -> C6H5NHCH2CH2OH
+ НCl. Чаще для синтеза этих же соед. применяют этиленоксид, леско реагирующий
с А. в присут. небольших кол-в Н2О:
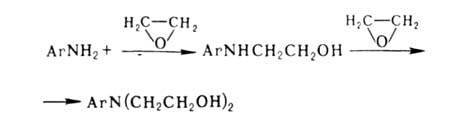
Поскольку скорости алкилирования на первой и второй стадиях одинаковы,
для получения моноалкильного производного этиленоксид берут в кол-ве, значительно
меньшем рассчитанного.
Цианэтильные производные первичных и вторичных А. получают р-цией их
с акрилонитрилом в присут. к-ты или щелочи: ArNH2 + CH2=CHCN
-> ArNHCH2CH2CN. Первичные и вторичные алифатич.
А. при взаимод. с CS2 образуют соли алкилдитиокарбаматов:
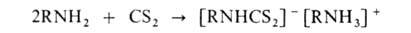
При нагр. первичных ароматич. А. с CS2 в присут. щелочи образуются
диарилтиомочевины, производные к-рых -важнейшие ускорители вулканизации:
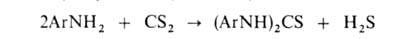
Гидрохлориды третичных А. при нагр. с разб. к-тами дезалкилируются:
(СН3)3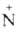 НСl
-> (CH3)2NH + СН3С1.
НСl
-> (CH3)2NH + СН3С1.
Под действием сильных окислителей, напр. КМnО4, первичные
алифатич. А. превращаются в смесь в-в, в к-рой преобладают альдегиды, первичные
ароматич. А. - в хиноны и их производные, вторичные алифатич. и ароматич.
А. - в тетразамещенные гидразины. При окислении третичных А. действием
Н2О2 или надкислот образуются N-оксиды А.:
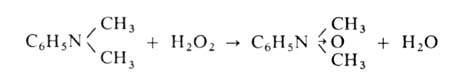
При замещении в ядро в ряду ароматич. А. аминогруппа ориентирует электроф.
замещение в орто-
и пара
-положения, а в сильно кислых средах
вследствие протонирования атома N - и в мета
-положение. Нитрование
первичных ароматич. А. осложняется их окислением, поэтому аминогруппу предварительно
ацилируют.
Осн. пром. методы синтеза А.:
1. Взаимод. спиртов с NH3 (аммонолиз спиртов) в присут. катализаторов
дегидратации (напр., А12О3, SiO2, ThO2,
алюмосиликатов, фосфатов металлов) при 300-500°С и 1-20 МПа. При этом образуются
смеси первичных, вторичных и третичных A.: ROH + NH3 -> RNH2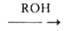 R2NH
R2NH RaN; одновременно происходит диспропорционирование A.: 2RNH2
-> R2NH + + NH3, R2NH + RNH2
-> R3N + NH3, 2R2NH -> R3N
+ RNH2. Этот метод применяется для произ-ва низших алифатич.
А., и прежде всего метил- и этиламинов. Синтез сопровождается образованием
значит. кол-в побочных продуктов - олефинов.
RaN; одновременно происходит диспропорционирование A.: 2RNH2
-> R2NH + + NH3, R2NH + RNH2
-> R3N + NH3, 2R2NH -> R3N
+ RNH2. Этот метод применяется для произ-ва низших алифатич.
А., и прежде всего метил- и этиламинов. Синтез сопровождается образованием
значит. кол-в побочных продуктов - олефинов.
При использовании вместо NH3 первичных или вторичных А. получаются
вторичные и (или) третичные А. Этот метод (аминолиз) распространен для
произ-ва N-алкил- и N,N-диалкиланилинов. Разработан аналогичный способ
получения анилина
взаимод. фенола с NH3. Очень легко
реагируют с NH3 нафтолы, образуя нафтиламины (см. Бухерера реакции
).
2. Восстановит. аминирование алифатич. и циклоалифатич. спиртов
в присут. Н2 на катализаторах гидрирования-дегидрирования (N1,
Со, Си, промотированное Fe). Процесс осуществляют при 150-250°С и 0,1-5
МПа:
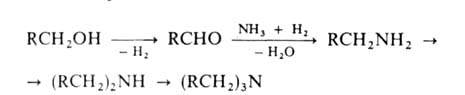
Вместо спиртов можно использовать альдегиды или кетоны; при этом образуется
смесь первичных и вторичных А.
3. Каталитич. гидрирование нитрилов (кат.-Ni или Со) при 100-130 °С
и 0,1-10 МПа. С хорошими выходами получаются первичные А. с примесью вторичных:
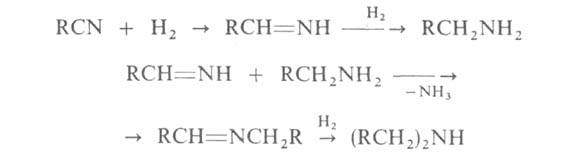
Этот метод применяется гл. обр. для произ-ва высших первичных и вторичных
алифатич. А. нормального строения.
4. Восстановление нитросоединений. Применяется гл. обр. для получения
первичных ароматич. A.: ArNO2 + 3Н2 -> ArNH2
+ 2Н2О. Наиб. распространено ката-литич. восстановление водородом
(кат.-Ni, Pt или Pd) в жидкой или паровой фазе, однако еще достаточно широко
в качестве восстановителей используются металлы (Fe или Zn) и сульфиды
щелочных металлов (см., напр., Зинина реакция
, Вешана реакции).
5. Р-ция амидов алифатич. и ароматич. карбоновых к-т со щелочными р-рами
С12, Вr2 или I2 с образованием первичных
А. При этом углеродная цепь укорачивается на один атом (Гофмана реакции
).
6. Р-ции с участием алкил- и арилгалогенидов. Конденсацией фталимида
К с алкилгалогенидами с послед. гидролизом (см. Габриеля реакция
)получают
чистые первичные алифатич. А.:
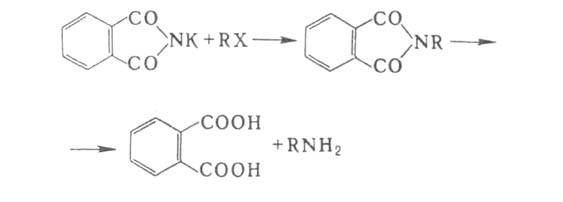
Арилгалогениды реагируют с NH3 и А. с трудом, поэтому в пром-сти
используют соед., в к-рых атом галогена активирован сильными электроноакцепторными
заместителями, чаще всего нитро- или сульфогруппами. Таким способом получают
разл. нитроанилины и производные дифениламина:
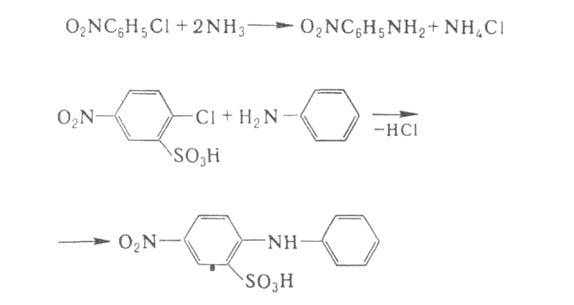
Аммонолиз и аминолиз арилгалогенидов с неактивированным галогеном требует
применения соед. Си в качестве катализаторов.
7. Нитрозирование N,N-диалкиланилинов с послед. гидролизом (получение
чистых вторичных алифатич. А.):
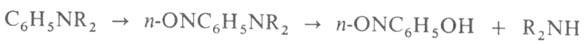
В лаборатории А. синтезируют, напр., р-циями Валлаха, Делепина, Курциуса,
Лейкарта, Лоссена, Манниха.
Специфич. р-ции обнаружения А.: первичные А. при нагр. с СНС13
в присут. щелочи превращаются в изонитрилы, обладающие очень сильным неприятным
запахом: RNH2 + СНС13 -> RNC + ЗНС1; при обработке
вторичных A. HNO2, послед. сплавлении полученного осадка с фенолом
и подкислении конц. H2SO4 появляется зеленое окрашивание.
Для идентификации первичных и вторичных А. используют их ацилирование в
амиды. Для количеств. определения А. применяют методы Кьельдаля и Ван Слайка
(для первичных алифатич. А.), бромометрич. метод, титрование р-рами к-т
в водной и неводной средах, газожидкостную хроматографию. Первичные ароматич.
А. определяют также фотометрически после образования соответствующих азосоединений
или оснований Шиффа.
А. - промежут. продукты в произ-ве красителей, пестицидов, полимеров
(в т.ч. полиамидов и полиуретанов), ингибиторов коррозии, ПАВ, флотореагентов,
абсорбентов, лек. ср-в (напр., сульфамидных препаратов), ускорителей вулканизации,
антиоксидантов и др.
Алифатич. А. поражают нервную систему, вызывают нарушения проницаемости
стенок кровеносных сосудов и клеточных мембран, ф-ций печени и развитие
дистрофии. Ароматич. А. вызывают образование метгемоглобина, угнетающего
центр. нервную систему. Нек-рые ароматич. А-канцерогены, вызывающие рак
мочевого пузыря у человека (напр., р-нафтиламин, бензидин, 4-аминобифенил).
Лит.: Терней А., Современная органическая химия, пер. с англ.,
т. 2, М., 1981; Общая органическая химия, пер. с англ., т. 3, М., 1982,
с. 11-91, 168-228; Ullmanns Encyklopadie, 4 Aufl., Bd 7, Weinheim, 1974:
Kirk-Othmer encyclopedia, 3 ed., v. 2, N.Y.-[a.o.], 1978, p. 272-376. Б.В.
Салол.


