АНИОННАЯ ПОЛИМЕРИЗАЦИЯ
, ионная полимеризация, при к-рой
концевое звено растущей цепи несет полный или частичный отрицат. заряд.
Традиционно к А. п. относят процессы, инициируемые соед. щелочных или щел.-зем.
металлов (либо своб. анионами). Процессы, развивающиеся с участием переходных
металлов, относят обычно, независимо от характера поляризации связи металл-углерод,
к координационно-ионной полимеризации
.
К А. п. способно большинство известных мономеров, напр. ненасыщенные
соед., содержащие в ос-положений электроноакцепторные группы (—СН=СН2,
—С6Н5, —COOR, —CN, —NO2 и др.), карбонильные
соед.,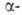 оксиды,
тиооксиды, лактоны, лактамы, силоксаны и др. гетероциклич. соединения.
Инициируется А.п. сильными основаниями, донорами электронов, электрич.
током и ионизирующими излучениями. Соед. щелочных и щел.-зем. металлов
(напр., орг. производные, алкоголяты, амиды) и др. в-ва основного характера
инициируют А.п. по типу кислотно-основного взаимод. (присоединение к мономеру
М инициатора АВ или его фрагмента А-):
оксиды,
тиооксиды, лактоны, лактамы, силоксаны и др. гетероциклич. соединения.
Инициируется А.п. сильными основаниями, донорами электронов, электрич.
током и ионизирующими излучениями. Соед. щелочных и щел.-зем. металлов
(напр., орг. производные, алкоголяты, амиды) и др. в-ва основного характера
инициируют А.п. по типу кислотно-основного взаимод. (присоединение к мономеру
М инициатора АВ или его фрагмента А-):
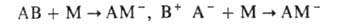
Своб. металлы, их ион-радикальные соли (напр., нафталинид Na) и др.
сильные доноры электронов действуют по типу окисления-восстановления (перенос
электрона к мономеру; Me-металл):
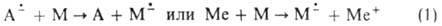
Процессы типа (1) характерны также для электрохим. и радиационно-хим.
инициирования. В инициировании по типу (1) участвуют промежут. ион-радикальные
частицы (М-), рекомбинация к-рых приводит к образованию молекул
с активными центрами на обоих концах: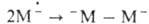 В принципе, при таком механизме возможно параллельное развитие анионных
и радикальных р-ций, однако в реально изученных системах случаи с заметным
участием последних не обнаружены.
В принципе, при таком механизме возможно параллельное развитие анионных
и радикальных р-ций, однако в реально изученных системах случаи с заметным
участием последних не обнаружены.
Активные центры А. п. в подавляющем большинстве случаев инициирования
щелочными, щел.-зем. металлами или их соединениями представляют собой производные
этих металлов. В зависимости от природы концевого мономерного звена (Р),
противоиона (Me+) и р-рителя (S)активные центры
могут существовать в виде различающихся по реакц. способности и стереоспецифичности
ковалентных поляризованных молекул (ф-ла II), их ассоциатов (I), ионных
пар разной степени сольватации (III, IV), своб. анионов Р (V):
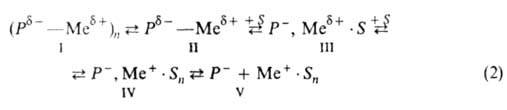
Как правило, противоион входит в состав активного центра и тем самым
оказывает непосредств. воздействие на акты роста цепи (исключение - полимеризация
на своб. анионах). В предельном случае присоединению мономера к растущей
цепи может предшествовать образование координац. комплекса с противоионом
(координационноионный механизм). Это создает большие возможности направленного
воздействия на полимеризацию и св-ва образующихся полимеров, чем в случае
процессов с участием "своб." частиц (своб. радикалов, анионов и катионов).
Для А. п. характерна, как правило, относит. стабильность активных центров.
В ряде случаев, напр. при А. п. неполярных мономеров в углеводородных р-рителях,
суммарный процесс включает практически лишь стадии инициирования и роста
цепи (р-ции обрыва и передачи цепи отсутствуют или идут с очень малыми
скоростями). При этом образуются т. наз. живущие полимеры, концевые группы
к-рых сохраняют способность к присоединению мономера или др. реагентов
и после завершения полимеризации. Такие полимеры - удобный объект как для
исследования механизма А. п., так и для решения разл. синтетич. задач:
получения полимеров с заданным ММР, в т.ч. практически монодисперсных;
синтеза полимеров и олигомеров с концевыми функц. группами, способными
к дальнейшим превращ. поликонденсац. или полимеризац. типа, а также блоксополимеров,
привитых сополимеров и разл. полимеров с регулируемым типом разветвления
и др.
А. п. мономеров с полярными функц. группами - более сложный процесс,
сопровождающийся дезактивацией активных центров при взаимод. с функц. группами
мономера и полимера. Энергия активации побочных р-ций (как и передачи цепи
на р-ритель в случае в-в с подвижным атомом Н, напр. толуола), как правило,
выше, чем энергия активации роста цепи; поэтому понижение т-ры полимеризации
способствует обычно подавлению побочных р-ций.
Скорость А. п., особенно при умеренных т-рах, в большинстве случаев
значительно выше скорости радикальной полимеризации. Это обычно связано
с более высокой действующей концентрацией активных частиц (в пределе она
м.б. равна исходной концентрации инициатора). Собственная же реакц. способность
разл. форм активных центров варьирует в очень широких пределах даже для
одного и того же мономера. Напр., для А. п. стирола при 30 °С порядок величины
абс. константы скорости роста цепи (в л/моль*с) при переходе вдоль равновесий
(2) изменяется от 10-1 (литиевые ассоциаты, II) до 105
(своб. анионы, V).
Общая кинетич. картина А. п. существенно осложнена упомянутой выше множественностью
форм существования активных центров. Помимо указанных в ур-ниях (2), в
ряде процессов играют роль и более сложные образования, напр. ионные тройники
типа Р -, Me+, Р- . Поэтому
даже в случае живущих полимеров при быстрой стадии инициирования, когда
суммарная концентрация растущих цепей равна исходной концентрации инициатора
(с0), общая скорость р-ции роста цепи (Vp)далеко
не всегда описывается простым ур-нием: Vp = kpc0
[Ml, где kp - константа скорости р-ции. Часто наблюдаются
более сложные зависимости общего вида:
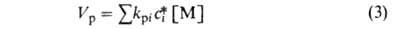
(сi* и kрi|-концентрация и константа скорости
роста i-того активного центра), учитывающие вклад разл. форм активных центров;
при этом суммарный порядок р-ции по инициатору варьирует от 1 до 0, а порядок
по мономеру равен в большинстве случаев 1. Наиб. важные частные случаи
ур-ния (3):
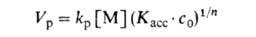
(рост на мономерной форме активных центров при концентрац. преобладании
малоактивных n-мерных ассоциатов; Касс-константа ассоциации)
и
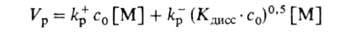
(одноврем. рост на своб. ионах и ионных парах при концентрац. преобладании
последних; Kдисс - константа диссоциации активных центров на
ионы).
Для строгой интерпретации кинетич. данных и расчета абс. значений элементарных
констант необходимо независимое определение Касс, Kдисc
и констант др. равновесий типа (2). В ряде случаев это можно осуществить
с помощью спектральных, кондуктометрич. и др. измерений, однако в целом
А.п. количественно значительно менее изучена, чем, напр., радикальная полимеризация.
Участие противоиона в актах роста цепи обусловливает большие возможности
воздействия на микроструктуру полимера, вплоть до образования в нек-рых
случаях стереорегулярных и оптически активных полимеров. В наиб. степени
ориентирующее влияние противоиона проявляется в углеводородной среде, где
в присут. Li, наиб. стереоспецифичного из щелочных металлов, образуются
1,4-полидиены (с преобладанием цис-структуры в случае изопрена или с равным
содержанием цис-
и транс-структур в случае бутадиена) и изотактич.
полиметилметакрилат. Среди щел.-зем. металлов образованию цис-1,4-полидиенов
и изотактич. полиметилметакрилата в наиб. степени способствует Ва. Электронодонорные
соед., насыщающие координац. сферу противоиона, благоприятствуют 1,2(3,4)-присоединению
диенов и образованию синдиотактич. полиметилметакрилата.
В пром-сти А. п. применяют гл. обр. для синтеза эластомерных материалов
(непрерывной полимеризацией в р-ре, преим. на литиевых инициаторах)- 1,4-
и 1,2-полибутадиена, статистич. сополимера бутадиена со стиролом, бутадиенстирольного
термоэластопласта; объем произ-ва этих полимеров составляет ок. 1 млн.
т/год. Методами А. п. синтезируют также олигомеры бутадиена с концевыми
функц. группами, поли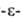 капроамид,
полиэтиленоксид, полиформальдегид, полисилоксаны и др. Осн. достоинства
А. п.-легкость управления, возможность получения почти всех перечисленных
гомо- и сополимеров бутадиена на одном и том же оборудовании при миним.
изменениях технол. процесса, наличие долгоживущих активных центров, высокая
чистота получаемых продуктов.
капроамид,
полиэтиленоксид, полиформальдегид, полисилоксаны и др. Осн. достоинства
А. п.-легкость управления, возможность получения почти всех перечисленных
гомо- и сополимеров бутадиена на одном и том же оборудовании при миним.
изменениях технол. процесса, наличие долгоживущих активных центров, высокая
чистота получаемых продуктов.
Систематич. изучение А. п. ненасыщенных соед. началось в 20-х гг. 20
в. (С. В. Лебедев, К. Циглер). Работы по теории А. п. и ее практич. реализации
особенно интенсивно стали развиваться с сер. 50-х гг., когда была открыта
способность Li вызывать образование цис-1,4-полиизопрена, близкого по структуре
и св-вам к НК, и были в полной мере осознаны синтетич. возможности живущих
полимеров.
Лит.: Шварц М., Анионная полимеризация, пер. с англ., М., 1971;
Ерусалимский Б. Л., Любе цк и и С. Г., Процессы ионной полимеризации, Л.,
1974; Арест-Якубович А. А., "Успехи химии", 1981, т. 50, в. 6, с. 1141-67.
А. А. Арест-Якубович.


